Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance



Abstract
1. Introduction
2. Known Resistance Mechanism to TKI
2.1. Mutations and Redundant Branches in Target Signalling Pathways
2.2. Role and Alterations of Transporter Proteins in TKI Resistance
2.3. Current Strategies to Overcome Resistance in TKI Based Therapy
2.4. Hurdles in Overcoming Resistance to TKI
3. Molecular Changes of Transporter Proteins in Drug Resistance
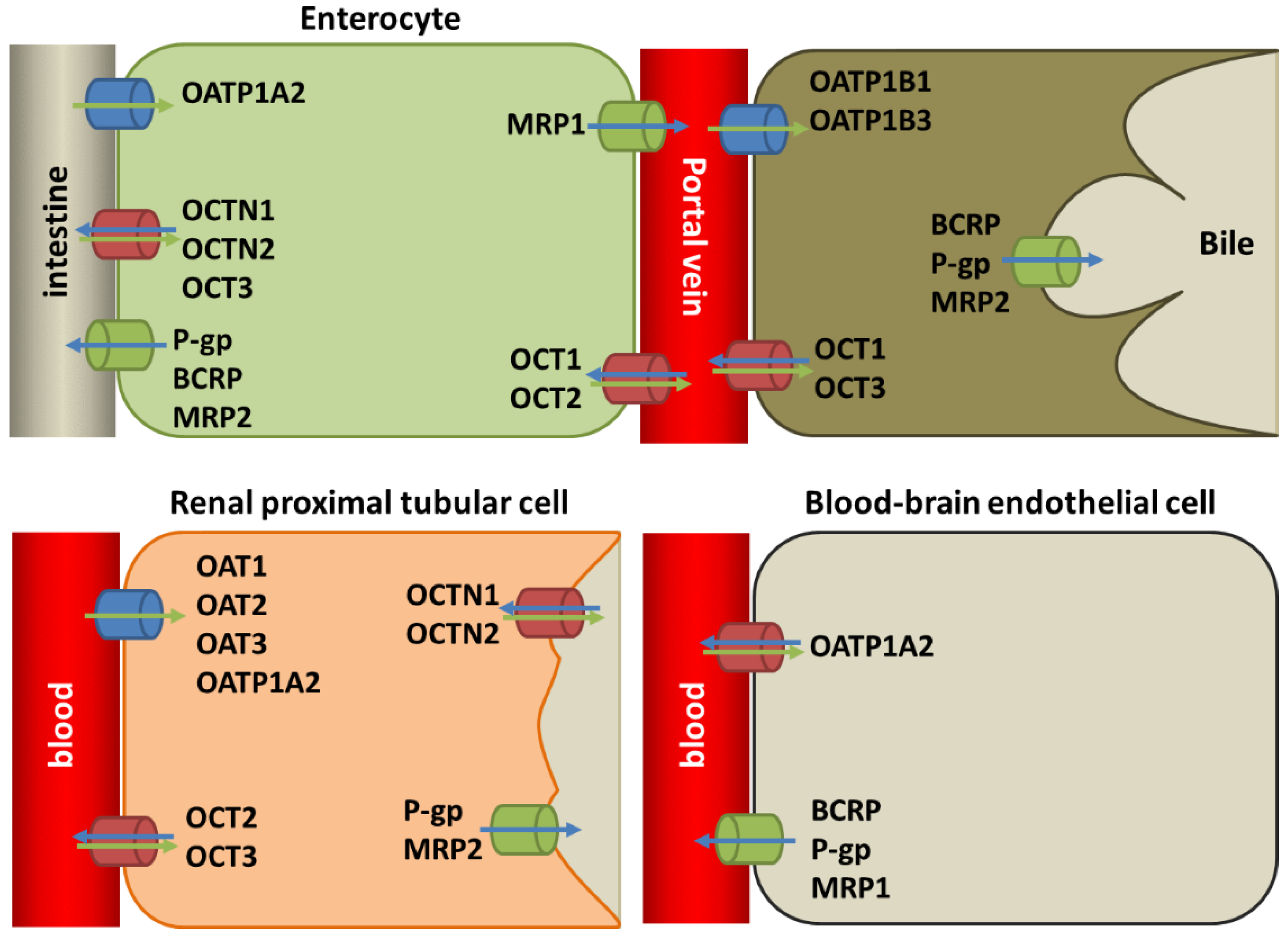
3.1. General Overview of the Transporters Involved in Cellular Uptake and Extrusion of TKIs
3.2. Transporter Proteins in ADME and TKI MDR in Cancer
3.3. Molecular Alterations in Transporter Proteins Affecting Drug Resistance to TKIs
3.3.1. Genetic Polymorphisms
3.3.2. Other Molecular Alterations Affecting Transporter Expression and Function
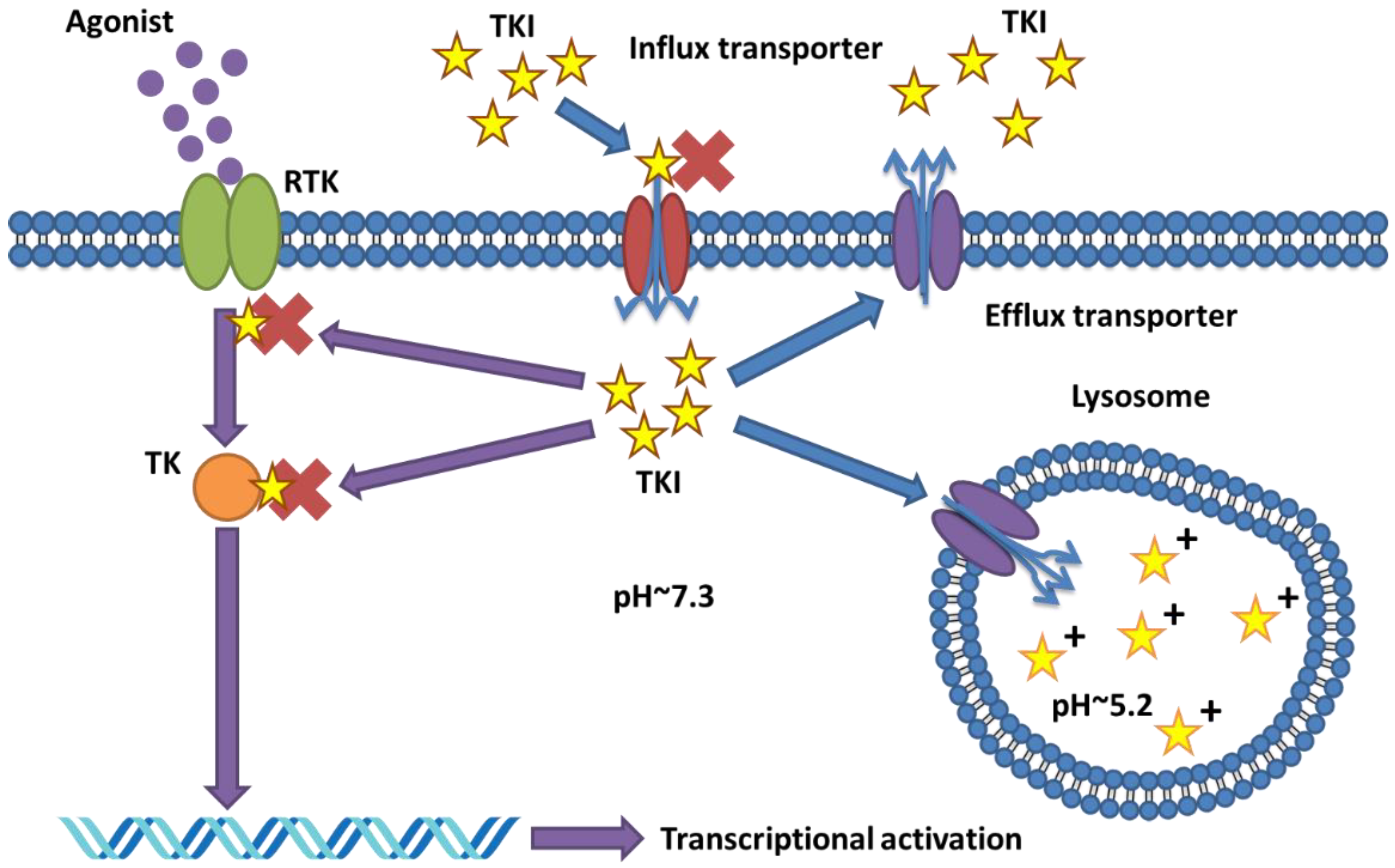
4. Lysosomal Sequestration of TKIs as Mechanism of Drug Resistance
4.1. Discovery of Lysosomal Sequestration of Protein Kinase Inhibitors
4.2. Mechanisms of Drug Sequestration and the Role of Transporters
5. Overcoming Transport- and Lysosome-Mediated (Multi) drug Resistance to TKIs
5.1. Overcoming Transport-Mediated (Multi)drug Resistance to TKIs
5.2. Overcoming Gut Epithelial Transport Mediated Resistance
5.3. Overcoming Lysosome-Mediated (Multi)drug Resistance to TKIs
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Talpaz, M.; Hehlmann, R.; Quintas-Cardama, A.; Mercer, J.; Cortes, J. Re-emergence of interferon-alpha in the treatment of chronic myeloid leukemia. Leukemia 2013, 27, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Xie, Q.; Zhang, Z.; Zhang, H. Protein kinase inhibitors for acute leukemia. Biomark. Res. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Miller, N. Durability of Kinase-Directed Therapies—A Network Perspective on Response and Resistance. Mol. Cancer Ther. 2015, 14, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Rethinking the war on cancer. Lancet 2014, 383, 558–563. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet. Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet. Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet. Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Gillis, N.K.; McLeod, H.L. The pharmacogenomics of drug resistance to protein kinase inhibitors. Drug Resist. Updates 2016, 28, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Neul, C.; Schaeffeler, E.; Sparreboom, A.; Laufer, S.; Schwab, M.; Nies, A.T. Impact of Membrane Drug Transporters on Resistance to Small-Molecule Tyrosine Kinase Inhibitors. Trends Pharmacol. Sci. 2016, 37, 904–932. [Google Scholar] [CrossRef] [PubMed]
- Ronaldson, P.T.; Bauer, B.; El-Kattan, A.F.; Shen, H.; Salphati, L.; Louie, S.W. Highlights From the American Association of Pharmaceutical Scientists/ International Transporter Consortium Joint Workshop on Drug Transporters in Absorption, Distribution, Metabolism, and Excretion: From the Bench to the Bedside—Clinical Pharmacology Considerations. Clin. Pharmacol. Ther. 2016, 100, 419–422. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal sequestration of hydrophobic weak base chemotherapeutics triggers lysosomal biogenesis and lysosome-dependent cancer multidrug resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLOS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Boil. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lin, Y.L.; Hsu, W.H.; Chen, H.Y.; Chang, Y.C.; Yu, C.J.; Shih, J.Y.; Lin, C.C.; Chen, K.Y.; Ho, C.C.; et al. Bcl-2-like protein 11 deletion polymorphism predicts survival in advanced non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Somwar, R.; Politi, K.; Balak, M.; Chmielecki, J.; Jiang, X.; Pao, W. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007, 4, e294. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Lee, S.S.; Jeong, S.H.; Ahn, J.S.; Yang, D.H.; Lee, J.J.; Shin, M.G.; Kim, H.J. OCT-1, ABCB1, and ABCG2 Expression in Imatinib-Resistant Chronic Myeloid Leukemia Treated with Dasatinib or Nilotinib. Chonnam Med. J. 2014, 50, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.B.; Hughes, T.P.; White, D.L. OCT1 and imatinib transport in CML: Is it clinically relevant? Leukemia 2015, 29, 1960–1969. [Google Scholar] [CrossRef]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin. Pharmacokinet. 2015, 54, 709–735. [Google Scholar] [CrossRef]
- Durmus, S.; Hendrikx, J.J.; Schinkel, A.H. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv. Cancer Res. 2015, 125, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Brok, M.; Wiemer, E.A.; de Bruijn, E.A.; Guetens, G.; de Boeck, G.; Sparreboom, A.; Verweij, J.; Nooter, K. Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Boil. Ther. 2005, 4, 747–752. [Google Scholar] [CrossRef]
- Bitencourt, R.; Zalcberg, I.; Louro, I.D. Imatinib resistance: A review of alternative inhibitors in chronic myeloid leukemia. Rev. Bras. Hematol. E Hemoter. 2011, 33, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, C.; Li, D.; Li, S. Molecular and cellular bases of chronic myeloid leukemia. Protein Cell 2010, 1, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.B.; Gunby, R.H.; Piazza, R.; Galietta, A.; Rostagno, R.; Scapozza, L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet. Oncol. 2003, 4, 75–85. [Google Scholar] [CrossRef]
- Hochhaus, A.; Kreil, S.; Corbin, A.S.; La Rosee, P.; Muller, M.C.; Lahaye, T.; Hanfstein, B.; Schoch, C.; Cross, N.C.; Berger, U.; et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002, 16, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet. Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Van Der Steen, N.; Caparello, C.; Rolfo, C.; Pauwels, P.; Peters, G.J.; Giovannetti, E. New developments in the management of non-small-cell lung cancer, focus on rociletinib: What went wrong? OncoTargets Ther. 2016, 9, 6065–6074. [Google Scholar] [CrossRef] [PubMed]
- Van Der Steen, N.; Giovannetti, E.; Carbone, D.; Leonetti, A.; Rolfo, C.D.; Peters, G. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer and strategies to overcome it. Cancer Drug Resist. 2018. [Google Scholar] [CrossRef]
- Nguyen, K.S.; Kobayashi, S.; Costa, D.B. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin. Lung Cancer 2009, 10, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsui, S.T.; Liu, C.; Song, Y.; Liu, D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 59. [Google Scholar] [CrossRef]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef]
- Muller, I.B.; De Langen, A.J.; Honeywell, R.J.; Giovannetti, E.; Peters, G.J. Overcoming crizotinib resistance in ALK-rearranged NSCLC with the second-generation ALK-inhibitor ceritinib. Expert Rev. Anticancer Ther. 2016, 16, 147–157. [Google Scholar] [CrossRef]
- Muller, I.B.; de Langen, A.J.; Giovannetti, E.; Peters, G.J. Anaplastic lymphoma kinase inhibition in metastatic non-small cell lung cancer: Clinical impact of alectinib. OncoTargets Ther. 2017, 10, 4535–4541. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.W.; Richardson, P.F.; Bailey, S.; Brooun, A.; Burke, B.J.; Collins, M.R.; Cui, J.J.; Deal, J.G.; Deng, Y.L.; Dinh, D.; et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sparidans, R.W.; Wang, Y.; Lebre, M.C.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (MDR1/ABCB1) restricts brain accumulation and cytochrome P450-3A (CYP3A) limits oral availability of the novel ALK/ROS1 inhibitor lorlatinib. Int. J. Cancer 2018, 143, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.; Muller, I.; Giovannetti, E. Should alectinib or ceritinib be given as first line therapy for ALK positive non-small cell lung cancer patients instead of crizotinib? Transl. Cancer Res. 2017, 6, S1010–S1013. [Google Scholar] [CrossRef]
- Peters, G.J.; van der Wilt, C.L.; van Moorsel, C.J.; Kroep, J.R.; Bergman, A.M.; Ackland, S.P. Basis for effective combination cancer chemotherapy with antimetabolites. Pharmacol. Ther. 2000, 87, 227–253. [Google Scholar] [CrossRef]
- Bijnsdorp, I.V.; Giovannetti, E.; Peters, G.J. Analysis of drug interactions. Methods Mol. Biol. 2011, 731, 421–434. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Weiss, A.; Ding, X.; Dyson, P.J.; van den Bergh, H.; Griffioen, A.W.; Ho, C.M. Optimization of drug combinations using Feedback System Control. Nat. Protoc. 2016, 11, 302–315. [Google Scholar] [CrossRef]
- He, M.; Wei, M.J. Reversing multidrug resistance by tyrosine kinase inhibitors. Chin. J. Cancer 2012, 31, 126–133. [Google Scholar] [CrossRef]
- Wang, F.; Chen, Y.; Huang, L.; Liu, T.; Huang, Y.; Zhao, J.; Wang, X.; Yang, K.; Ma, S.; Huang, L.; et al. Cetuximab enhanced the efficacy of chemotherapeutic agent in ABCB1/P-glycoprotein-overexpressing cancer cells. OncoTarget 2015, 6, 40850–40865. [Google Scholar] [CrossRef]
- Deng, J.; Shao, J.; Markowitz, J.S.; An, G. ABC transporters in multi-drug resistance and ADME-Tox of small molecule tyrosine kinase inhibitors. Pharm. Res. 2014, 31, 2237–2255. [Google Scholar] [CrossRef]
- Anreddy, N.; Gupta, P.; Kathawala, R.J.; Patel, A.; Wurpel, J.N.; Chen, Z.S. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules 2014, 19, 13848–13877. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.G.; Honeywell, R.J.; Dekker, H.; Peters, G.J. Physicochemical properties of novel protein kinase inhibitors in relation to their substrate specificity for drug transporters. Expert Opin. Drug Metab. Toxicol. 2015, 11, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Zheng, F.; Ren, D.; Du, F.; Dong, Q.; Wang, Z.; Zhao, F.; Ahmad, R.; Zhao, J. Anlotinib: A novel multi-targeting tyrosine kinase inhibitor in clinical development. J. Hematol. Oncol. 2018, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wu, Y.L. Clinical trials of tyrosine kinase inhibitors for lung cancer in China: A review. J. Hematol. Oncol. 2017, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Broekman, F.; Giovannetti, E.; Peters, G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011, 2, 80–93. [Google Scholar] [CrossRef]
- Genentech. ALECENSA® (alectinib): Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf (accessed on 6 August 2018).
- Redaelli, S.; Perini, P.; Ceccon, M.; Piazza, R.; Rigolio, R.; Mauri, M.; Boschelli, F.; Giannoudis, A.; Gambacorti-Passerini, C. In vitro and in vivo identification of ABCB1 as an efflux transporter of bosutinib. J. Hematol. Oncol. 2015, 8, 81. [Google Scholar] [CrossRef]
- ALUNBRIGTM (Brigatinib) Tablets, f.o.u. ALUNBRIGTM (Brigatinib) Tablets, for Oral Use 2017. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm555841.htm (accessed on 28 April 2017).
- Exelixis, I. CABOMETYX® (cabozantinib): Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208692s002lbl.pdf (accessed on 6 August 2018).
- Corporation, N.P. ZYKADIA® (ceritinib): Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/205755s010lbl.pdf (accessed on 6 August 2018).
- Weiss, J.; Theile, D.; Dvorak, Z.; Haefeli, W.E. Interaction potential of the multitargeted receptor tyrosine kinase inhibitor dovitinib with drug transporters and drug metabolising enzymes assessed in vitro. Pharmaceutics 2014, 6, 632–650. [Google Scholar] [CrossRef]
- Eisai Manufacturing Ltd. Kisplyx (lenvatinib): Summary of Product Characteristics. Available online: https://ec.europa.eu/health/documents/community-register/2017/20170227137185/anx_137185_en.pdf (accessed on 7 August 2018).
- Boehringer Ingelheim Pharma GmbH & Co. KG. Vargatef (nintedanib): Summary of Product Characteristics. Available online: https://www.ema.europa.eu/documents/product-information/vargatef-epar-product-information_en.pdf (accessed on 7 August 2018).
- AB, A. Tagrisso (osimertinib): Summary of Product Characteristics. Available online: https://www.ema.europa.eu/documents/product-information/tagrisso-epar-product-information_en.pdf (accessed on 7 August 2018).
- Corporation, I. JAKAFI™ (ruxolitinib): Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf (accessed on 7 August 2018).
- Harrach, S.; Edemir, B.; Schmidt-Lauber, C.; Pap, T.; Bertrand, J.; Ciarimboli, G. Importance of the novel organic cation transporter 1 for tyrosine kinase inhibition by saracatinib in rheumatoid arthritis synovial fibroblasts. Sci. Rep. 2017, 7, 1258. [Google Scholar] [CrossRef]
- de Gooijer, M.C.; Zhang, P.; Weijer, R.; Buil, L.C.M.; Beijnen, J.H.; van Tellingen, O. The impact of P-glycoprotein and breast cancer resistance protein on the brain pharmacokinetics and pharmacodynamics of a panel of MEK inhibitors. Int. J. Cancer 2018, 142, 381–391. [Google Scholar] [CrossRef]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef]
- Yang, J.J.; Milton, M.N.; Yu, S.; Liao, M.; Liu, N.; Wu, J.T.; Gan, L.; Balani, S.K.; Lee, F.W.; Prakash, S.; et al. P-glycoprotein and breast cancer resistance protein affect disposition of tandutinib, a tyrosine kinase inhibitor. Drug Metab. Lett. 2010, 4, 201–212. [Google Scholar] [CrossRef]
- Pharma, N. Mekinist (trametinib): Summary of Product Characteristics. Available online: https://www.ema.europa.eu/documents/product-information/mekinist-epar-product-information_en.pdf (accessed on 8 August 2018).
- Pfizer, I. XELJANZ® (tofacitinib): Highlights of Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203214s017,208246s003lbl.pdf (accessed on 8 August 2018).
- Kovacsics, D.; Patik, I.; Ozvegy-Laczka, C. The role of organic anion transporting polypeptides in drug absorption, distribution, excretion and drug-drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Meier, P.J.; Ballatori, N. Oatp2 mediates bidirectional organic solute transport: A role for intracellular glutathione. Mol. Pharmacol. 2000, 58, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Satlin, L.M.; Amin, V.; Wolkoff, A.W. Organic anion transporting polypeptide mediates organic anion/HCO3- exchange. J. Boil. Chem. 1997, 272, 26340–26345. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Asp. Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [PubMed]
- Ballestero, M.R.; Monte, M.J.; Briz, O.; Jimenez, F.; Gonzalez-San Martin, F.; Marin, J.J. Expression of transporters potentially involved in the targeting of cytostatic bile acid derivatives to colon cancer and polyps. Biochem. Pharmacol. 2006, 72, 729–738. [Google Scholar] [CrossRef]
- Bronger, H.; Konig, J.; Kopplow, K.; Steiner, H.H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef]
- Liedauer, R.; Svoboda, M.; Wlcek, K.; Arrich, F.; Ja, W.; Toma, C.; Thalhammer, T. Different expression patterns of organic anion transporting polypeptides in osteosarcomas, bone metastases and aneurysmal bone cysts. Oncol. Rep. 2009, 22, 1485–1492. [Google Scholar]
- Arakawa, H.; Nakanishi, T.; Yanagihara, C.; Nishimoto, T.; Wakayama, T.; Mizokami, A.; Namiki, M.; Kawai, K.; Tamai, I. Enhanced expression of organic anion transporting polypeptides (OATPs) in androgen receptor-positive prostate cancer cells: Possible role of OATP1A2 in adaptive cell growth under androgen-depleted conditions. Biochem. Pharmacol. 2012, 84, 1070–1077. [Google Scholar] [CrossRef]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef]
- Thakkar, N.; Lockhart, A.C.; Lee, W. Role of Organic Anion-Transporting Polypeptides (OATPs) in Cancer Therapy. AAPS J. 2015, 17, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Furihata, T.; Sun, Y.; Chiba, K. Cancer-type Organic Anion Transporting Polypeptide 1B3: Current Knowledge of the Gene Structure, Expression Profile, Functional Implications and Future Perspectives. Curr. Drug Metab. 2015, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Hosoyamada, M.; Sekine, T.; Kanai, Y.; Endou, H. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am. J. Physiol. 1999, 276, F122–F128. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar] [PubMed]
- Takeda, M.; Noshiro, R.; Onozato, M.L.; Tojo, A.; Hasannejad, H.; Huang, X.L.; Narikawa, S.; Endou, H. Evidence for a role of human organic anion transporters in the muscular side effects of HMG-CoA reductase inhibitors. Eur. J. Pharmacol. 2004, 483, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wu, R.R.; van Poelje, P.D.; Erion, M.D. Isolation of a family of organic anion transporters from human liver and kidney. Biochem. Biophys. Res. Commun. 2001, 283, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorf, C.; Ahlin, G.; Seithel, A.; Artursson, P.; Ungell, A.L.; Karlsson, J. Expression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab. Dispos. 2007, 35, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Shimoda, M.; Narikawa, S.; Kobayashi, Y.; Kobayashi, Y.; Yamamoto, T.; Sekine, T.; Cha, S.H.; Niwa, T.; et al. Interaction of human organic anion transporters 2 and 4 with organic anion transport inhibitors. J. Pharmacol. Exp. Ther. 2002, 301, 797–802. [Google Scholar] [CrossRef]
- Cha, S.H.; Sekine, T.; Fukushima, J.I.; Kanai, Y.; Kobayashi, Y.; Goya, T.; Endou, H. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol. Pharmacol. 2001, 59, 1277–1286. [Google Scholar] [CrossRef]
- Asif, A.R.; Steffgen, J.; Metten, M.; Grunewald, R.W.; Muller, G.A.; Bahn, A.; Burckhardt, G.; Hagos, Y. Presence of organic anion transporters 3 (OAT3) and 4 (OAT4) in human adrenocortical cells. Pflugers Arch. 2005, 450, 88–95. [Google Scholar] [CrossRef]
- Barendt, W.M.; Wright, S.H. The human organic cation transporter (hOCT2) recognizes the degree of substrate ionization. J. Boil. Chem. 2002, 277, 22491–22496. [Google Scholar] [CrossRef]
- Gorboulev, V.; Ulzheimer, J.C.; Akhoundova, A.; Ulzheimer-Teuber, I.; Karbach, U.; Quester, S.; Baumann, C.; Lang, F.; Busch, A.E.; Koepsell, H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Boil. 1997, 16, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, W.; Ganapathy, M.E.; Wang, H.; Kekuda, R.; Conway, S.J.; Leibach, F.H.; Ganapathy, V. Structure, function, and regional distribution of the organic cation transporter OCT3 in the kidney. Am. J. Physiology. Ren. Physiol. 2000, 279, F449–F458. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Herrmann, E.; Brom, M.; Keppler, D. Vectorial transport of the plant alkaloid berberine by double-transfected cells expressing the human organic cation transporter 1 (OCT1, SLC22A1) and the efflux pump MDR1 P-glycoprotein (ABCB1). Naunyn-Schmiedeberg's Arch. Pharmacol. 2008, 376, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Lips, K.S.; Volk, C.; Schmitt, B.M.; Pfeil, U.; Arndt, P.; Miska, D.; Ermert, L.; Kummer, W.; Koepsell, H. Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am. J. Respir. Cell Mol. Boil. 2005, 33, 79–88. [Google Scholar] [CrossRef]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef]
- Muller, J.; Lips, K.S.; Metzner, L.; Neubert, R.H.; Koepsell, H.; Brandsch, M. Drug specificity and intestinal membrane localization of human organic cation transporters (OCT). Biochem. Pharmacol. 2005, 70, 1851–1860. [Google Scholar] [CrossRef]
- Muller, J.; Keiser, M.; Drozdzik, M.; Oswald, S. Expression, regulation and function of intestinal drug transporters: An update. Boil. Chem. 2017, 398, 175–192. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport--an update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Keppler, D. Progress in the Molecular Characterization of Hepatobiliary Transporters. Dig. Dis. 2017, 35, 197–202. [Google Scholar] [CrossRef]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef] [PubMed]
- Bakos, E.; Homolya, L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1). Pflugers Arch. 2007, 453, 621–641. [Google Scholar] [CrossRef]
- Nies, A.T.; Keppler, D. The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch. Eur. J. Physiol. 2007, 453, 643–659. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.V.; Iyer, S. Preclinical Pharmacokinetic Considerations for the Development of Antibody Drug Conjugates. Pharm. Res. 2015, 32, 3470–3479. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Koike, S.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Fujita, N. Dofequidar fumarate sensitizes cancer stem-like side population cells to chemotherapeutic drugs by inhibiting ABCG2/BCRP-mediated drug export. Cancer Sci. 2009, 100, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Lemos, C.; Jansen, G.; Peters, G.J. Drug transporters: Recent advances concerning BCRP and tyrosine kinase inhibitors. Br. J. Cancer 2008, 98, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Dessilly, G.; Elens, L.; Panin, N.; Karmani, L.; Demoulin, J.B.; Haufroid, V. ABCB1 1199G > A polymorphism (rs2229109) affects the transport of imatinib, nilotinib and dasatinib. Pharmacogenomics 2016, 17, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.T.; Soranzo, N.; Nimmo, E.R.; Tenesa, A.; Goldstein, D.B.; Satsangi, J. ABCB1/MDR1 gene determines susceptibility and phenotype in ulcerative colitis: Discrimination of critical variants using a gene-wide haplotype tagging approach. Hum. Mol. Genet. 2006, 15, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Dessilly, G.; Panin, N.; Elens, L.; Haufroid, V.; Demoulin, J.B. Impact of ABCB1 1236C > T-2677G > T-3435C > T polymorphisms on the anti-proliferative activity of imatinib, nilotinib, dasatinib and ponatinib. Sci. Rep. 2016, 6, 29559. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, K.; Boiso Moreno, S.; Jonsson, J.I.; Vikingsson, S.; Carlsson, B.; Green, H. Single-nucleotide polymorphisms of ABCG2 increase the efficacy of tyrosine kinase inhibitors in the K562 chronic myeloid leukemia cell line. Pharm. Genom. 2014, 24, 52–61. [Google Scholar] [CrossRef]
- Chen, X.; Chen, D.; Yang, S.; Ma, R.; Pan, Y.; Li, X.; Ma, S. Impact of ABCG2 polymorphisms on the clinical outcome of TKIs therapy in Chinese advanced non-small-cell lung cancer patients. Cancer Cell Int. 2015, 15, 43. [Google Scholar] [CrossRef]
- Noguchi, K.; Katayama, K.; Sugimoto, Y. Human ABC transporter ABCG2/BCRP expression in chemoresistance: Basic and clinical perspectives for molecular cancer therapeutics. Pharm. Pers. Med. 2014, 7, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Aozasa, N.; Kotani, H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int. J. Cancer 2004, 109, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kondo, C.; Suzuki, H.; Itoda, M.; Ozawa, S.; Sawada, J.; Kobayashi, D.; Ieiri, I.; Mine, K.; Ohtsubo, K.; Sugiyama, Y. Functional analysis of SNPs variants of BCRP/ABCG2. Pharm. Res. 2004, 21, 1895–1903. [Google Scholar] [CrossRef]
- Poonkuzhali, B.; Lamba, J.; Strom, S.; Sparreboom, A.; Thummel, K.; Watkins, P.; Schuetz, E. Association of breast cancer resistance protein/ABCG2 phenotypes and novel promoter and intron 1 single nucleotide polymorphisms. Drug Metab. Dispos. 2008, 36, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, A.; Wang, L.; Jorgensen, A.L.; Xinarianos, G.; Davies, A.; Pushpakom, S.; Liloglou, T.; Zhang, J.E.; Austin, G.; Holyoake, T.L.; et al. The hOCT1 SNPs M420del and M408V alter imatinib uptake and M420del modifies clinical outcome in imatinib-treated chronic myeloid leukemia. Blood 2013, 121, 628–637. [Google Scholar] [CrossRef]
- Scotto, K.W. Transcriptional regulation of ABC drug transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef]
- Nagalingam, A.; Tighiouart, M.; Ryden, L.; Joseph, L.; Landberg, G.; Saxena, N.K.; Sharma, D. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis 2012, 33, 918–930. [Google Scholar] [CrossRef]
- Ogretmen, B.; Safa, A.R. Identification and characterization of the MDR1 promoter-enhancing factor 1 (MEF1) in the multidrug resistant HL60/VCR human acute myeloid leukemia cell line. Biochemistry 2000, 39, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett 2016, 370, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, H.; Liu, X.; Evans, B.R.; Medina, D.J.; Liu, C.G.; Yang, J.M. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem. Pharmacol. 2008, 76, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Tomiyasu, H.; Goto-Koshino, Y.; Fujino, Y.; Ohno, K.; Tsujimoto, H. Epigenetic regulation of the ABCB1 gene in drug-sensitive and drug-resistant lymphoid tumour cell lines obtained from canine patients. Vet. J. 2014, 199, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Gump, J.L.; Zhang, C.; Cook, J.M.; Marchion, D.; Hazlehurst, L.; Munster, P.; Schell, M.J.; Dalton, W.S.; Sullivan, D.M. ABCG2 expression, function, and promoter methylation in human multiple myeloma. Blood 2006, 108, 3881–3889. [Google Scholar] [CrossRef]
- Kim, D.H.; Sriharsha, L.; Xu, W.; Kamel-Reid, S.; Liu, X.; Siminovitch, K.; Messner, H.A.; Lipton, J.H. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin. Cancer Res. 2009, 15, 4750–4758. [Google Scholar] [CrossRef]
- Smith, P.J.; Sykes, H.R.; Fox, M.E.; Furlong, I.J. Subcellular distribution of the anticancer drug mitoxantrone in human and drug-resistant murine cells analyzed by flow cytometry and confocal microscopy and its relationship to the induction of DNA damage. Cancer Res. 1992, 52, 4000–4008. [Google Scholar]
- Duvvuri, M.; Gong, Y.; Chatterji, D.; Krise, J.P. Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60. J. Boil. Chem. 2004, 279, 32367–32372. [Google Scholar] [CrossRef]
- Kaufmann, A.M.; Krise, J.P. Lysosomal sequestration of amine-containing drugs: Analysis and therapeutic implications. J. Pharm. Sci. 2007, 96, 729–746. [Google Scholar] [CrossRef]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef]
- Halaby, R. Role of lysosomes in cancer therapy. Res. Rep. Boil. 2015, 6, 147–155. [Google Scholar] [CrossRef]
- Mrschtik, M.; Ryan, K.M. Lysosomal proteins in cell death and autophagy. FEBS J. 2015, 282, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Gotink, K.J.; Verheul, H.M. The Potential Role of Lysosomal Sequestration in Sunitinib Resistance of Renal Cell Cancer. J. Kidney Cancer VHL 2015, 2, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J. Boil. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Koch, R.; Radunski, U.; Corsham, S.; Cheong, N.; Inagaki, N.; Ban, N.; Wenzel, D.; Reinhardt, D.; Zapf, A.; et al. Intracellular ABC transporter A3 confers multidrug resistance in leukemia cells by lysosomal drug sequestration. Leukemia 2008, 22, 1576–1586. [Google Scholar] [CrossRef]
- Ferrao, P.; Sincock, P.; Cole, S.; Ashman, L. Intracellular P-gp contributes to functional drug efflux and resistance in acute myeloid leukaemia. Leuk. Res. 2001, 25, 395–405. [Google Scholar] [CrossRef]
- Guo, X.; To, K.K.W.; Chen, Z.; Wang, X.; Zhang, J.; Luo, M.; Wang, F.; Yan, S.; Fu, L. Dacomitinib potentiates the efficacy of conventional chemotherapeutic agents via inhibiting the drug efflux function of ABCG2 in vitro and in vivo. J. Exp. Clin. Cancer Res. 2018, 37, 31. [Google Scholar] [CrossRef]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.F.; Zhang, W.; Zeng, L.; Lei, Z.N.; Cai, C.Y.; Gupta, P.; Yang, D.H.; Cui, Q.; Qin, Z.D.; Chen, Z.S.; et al. Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett. 2018, 421, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, A.C.; Al Saig, F.; Cloos, J.; Jansen, G.; Peters, G.J. How to overcome ATP-binding cassette drug efflux transporter-mediated drug resistance? Cancer Drug Resist. 2018, 1, 6–29. [Google Scholar] [CrossRef]
- Marcelletti, J.F.; Sikic, B.I.; Cripe, L.D.; Paietta, E. Evidence of a role for functional heterogeneity in multidrug resistance transporters in clinical trials of P-glycoprotein modulation in acute myeloid leukemia. Cytom. Part B Clin. Cytom. 2018. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhang, Y.K.; Kathawala, R.J.; Chen, Z.S. Repositioning of Tyrosine Kinase Inhibitors as Antagonists of ATP-Binding Cassette Transporters in Anticancer Drug Resistance. Cancers 2014, 6, 1925–1952. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Abuznait, A.H.; Singh, S.; Xiao, Z.J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, M.; Wang, F.; Zhen, C.; Luo, M.; Fang, X.; Zhang, H.; Zhang, J.; Li, Q.; Fu, L. Ceritinib Enhances the Efficacy of Substrate Chemotherapeutic Agent in Human ABCB1-Overexpressing Leukemia Cells In Vitro, In Vivo and Ex-Vivo. Cell. Physiol. Biochem. 2018, 46, 2487–2499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Guo, X.; To, K.K.W.; Chen, Z.; Fang, X.; Luo, M.; Ma, C.; Xu, J.; Yan, S.; Fu, L. Olmutinib (HM61713) reversed multidrug resistance by inhibiting the activity of ATP-binding cassette subfamily G member 2 in vitro and in vivo. Acta Pharm. Sinica. B 2018, 8, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Honeywell, R.J.; Fatmawati, C.; Buddha, M.; Hitzerd, S.; Kathman, I.; Peters, G.J. Adaptation of a human gut epithelial model in relation to the assessment of clinical pharmacokinetic parameters for selected tyrosine kinase inhibitors. ADMET DMPK 2015, 3, 51–67. [Google Scholar] [CrossRef]
- Willemsen, A.E.; Lubberman, F.J.; Tol, J.; Gerritsen, W.R.; van Herpen, C.M.; van Erp, N.P. Effect of food and acid-reducing agents on the absorption of oral targeted therapies in solid tumors. Drug Discov. Today 2016, 21, 962–976. [Google Scholar] [CrossRef]
- Honeywell, R.J.; Hitzerd, S.; Kathmann, I.; Peters, G.J. Transport of six tyrosine kinase inhibitors: Active or passive? ADMET DMPK 2016, 4, 23–34. [Google Scholar] [CrossRef]
- Logan, R.; Funk, R.S.; Axcell, E.; Krise, J.P. Drug-drug interactions involving lysosomes: Mechanisms and potential clinical implications. Expert Opin. Drug Metab. Toxicol. 2012, 8, 943–958. [Google Scholar] [CrossRef]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 45–54. [Google Scholar] [CrossRef]
- Kallifatidis, G.; Hoepfner, D.; Jaeg, T.; Guzman, E.A.; Wright, A.E. The marine natural product manzamine A targets vacuolar ATPases and inhibits autophagy in pancreatic cancer cells. Mar. Drugs 2013, 11, 3500–3516. [Google Scholar] [CrossRef]
- Englinger, B.; Kallus, S.; Senkiv, J.; Heilos, D.; Gabler, L.; van Schoonhoven, S.; Terenzi, A.; Moser, P.; Pirker, C.; Timelthaler, G.; et al. Intrinsic fluorescence of the clinically approved multikinase inhibitor nintedanib reveals lysosomal sequestration as resistance mechanism in FGFR-driven lung cancer. J. Exp. Clin. Cancer Res. 2017, 36, 122. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Lacconi, V.; Peddis, M.; Lotti, L.V.; Di Renzo, L.; Gonnella, R.; Santarelli, R.; Trivedi, P.; Frati, L.; D'Orazi, G.; et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. 2013, 4, e730. [Google Scholar] [CrossRef] [PubMed]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Weiss, A.; van Beijnum, J.R.; Wong, T.J.; Kilarski, W.W.; Szewczyk, G.; Verheul, H.M.; Sarna, T.; van den Bergh, H.; Griffioen, A.W. Photoactivation of lysosomally sequestered sunitinib after angiostatic treatment causes vascular occlusion and enhances tumor growth inhibition. Cell Death Dis. 2015, 6, e1641. [Google Scholar] [CrossRef] [PubMed]
- Honeywell, R.J.; Hitzerd, S.; Kathmann, G.A.M.; Peters, G.J. Subcellular localization of several structurally different tyrosine kinase inhibitors. ADMET DMPK 2018, 6, 258–266. [Google Scholar] [CrossRef]
- Peters, G.J.; Kathman, I.; Jansen, G.; Valko, K.; Honeywell, R.J. Distribution of tyrosine kinase inhibitors in relation to biomimetic properties. In Proceedings of the 7th IAPC Meeting, Osaka, Japan, 28–30 August 2018; Abstract I-04. p. 5. [Google Scholar]
- Valko, K.; Teague, S.; Pidgeon, C. In vitro membrane binding and protein binding (IAM MB/PB technology) to estimate in vivo distribution: Applications in early drug discovery. ADMET DMPK 2017, 5, 14–38. [Google Scholar] [CrossRef]
- Lin, L.C.; Kuo, T.T.; Chang, H.Y.; Liu, W.S.; Hsia, S.M.; Huang, T.C. Manzamine A Exerts Anticancer Activity against Human Colorectal Cancer Cells. Mar. Drugs 2018, 16, 252. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Da Silva, C.G.; Peters, G.J.; Ossendorp, F.; Cruz, L.J. The potential of multi-compound naoparticles to bypass drug resistance in cancer. Cancer Chemother. Pharmacol. 2017, 80, 881–894. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drugs | Molecular Structure (SMILES) 1 | pKa/pKb (Predicted ChemAxon) | LogP (Predicted by ChemAxon) | Physiological Charge | logP | Molecular Target | Transporter Subtrate | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OATP1A2/1B1/1B3 | OAT1/2/3 | OCT1/2/3 | OCTN1/2 | P-gp | BCRP | MRP1/2 | ||||||||
| Afatinib | CN(C)C\C=C\C(=O)NC1=C(O[C@H]2CCOC2)C=C2N=CN=C(NC3=CC(Cl)=C(F)C=C3)C2=C1 | 12.49/8.81 | 3.76 | 1 | 3.59 | EGFRmut, ERBB2/4 | − | + | + | [12] | ||||
| Alectinib | CCC1=CC2=C(C=C1N1CCC(CC1)N1CCOCC1)C(C)(C)C1=C(C3=C(N1)C=C(C=C3)C#N)C2=O | 12.18/6.97 | 5.59 | 1 | 4.15 | ALK | */−/− | − | − | [56] | ||||
| Allitinib | C=CC(NC1=CC2=C(NC3=CC=C(OCC4=CC=CC(F)=C4)C(Cl)=C3)N=CN=C2C=C1)=O | 14.45/3.99 | 5.77 | 0 | EGFR1, ErbB2 (HER2) | |||||||||
| Anlotinib | COC1=CC2=C(OC3=CC=C4NC(C)=CC4=C3F)C=CN=C2C=C1OCC1(N)CC1 | 16.65/9.39 | 3.35 | 1 | VEGFR1/2/3, FGFR1/2/3/4, PDGFRα/ß,cKIT | |||||||||
| Apatinib | CC1=C([NH2+]C2=C(C(NC3=CC=C(C4(CCCC4)C#N)C=C3)=O)C=CC=N2)C=NC=C1.CS([O-])(=O)=O | no predicted data available | 1 | VEGFR,ROS-1,RET | + | + | ||||||||
| Avitinib | CN1CCN(C2=C(F)C=C(NC3=NC(OC4=CC(NC(C=C)=O)=CC=C4)=C(C=CN5)C5=N3)C=C2)CC | 12.6/7.25 | 3.88 | 0 | EGFRWT,T790M | |||||||||
| Axitinib | [H]\C(=C(\[H])C1=CC=CC=N1)C1=C2C=CC(SC3=CC=CC=C3C(O)=NC)=CC2=NN1 | 7.73/5.14 | 5.01 | 1 | 4.2 | VEGF, PDGF | */+/+ | + | − | [12,52] | ||||
| AZD3759 | COC(C(OC(N1CCN(C)C[C@H]1C)=O)=C2)=CC3=C2C(NC4=CC=CC(Cl)=C4F)=NC=N3 | 13.81/−7.1 | 4.03 | 0 | EGFR | |||||||||
| BGB-283 | O=C1NC2=NC=CC(OC3=CC([C@]4([H])[C@]([C@@H]4[C@@H]5NC6=CC=C(C(F)(F)F)C=C6N5)([H])O7)=C7C=C3)=C2CC1 | no predicted data available | 0 | BRAF, EGFR | ||||||||||
| Bosutinib | COC1=CC(NC2=C(C=NC3=CC(OCCCN4CCN(C)CC4)=C(OC)C=C23)C#N)=C(Cl)C=C1Cl | 15.48/8.43 | 4.09 | 1 | 5.4 | Bcr-Abl, Src | + | [57] | ||||||
| Brigatinib | COC1=CC(=CC=C1NC1=NC=C(Cl)C(NC2=CC=CC=C2P(C)(C)=O)=N1)N1CCC(CC1)N1CCN(C)CC1 | 12.88/8.54 | 3.66 | 1 | 4.6 | ALK, EGFR | */−/− | −/*/− | −/−/* | + | + | [58] | ||
| Cabozantinib | COC1=CC2=C(C=C1OC)C(OC1=CC=C(NC(=O)C3(CC3)C(=O)NC3=CC=C(F)C=C3)C=C1)=CC=N2 | 13.46/5.9 | 4.66 | 0 | 5.4 | c-Met, VEGFR2, AXL, RET | /+ | [59] | ||||||
| Canertinib | FC1=C(Cl)C=C(NC2=NC=NC3=CC(OCCCN4CCOCC4)=C(NC(=O)C=C)C=C23)C=C1 | 12.54/6.87 | 3.9 | 0 | 3.65 | EGFR | + | [48] | ||||||
| Capmatinib (INC280) | O=C(NC)C1=CC=C(C2=NN3C(N=C2)=NC=C3CC4=CC=C5N=CC=CC5=C4)C=C1F | 12.77/4.55 | 2.96 | 0 | c-Met | |||||||||
| Ceritinib | CC(C)OC1=C(NC2=NC=C(Cl)C(N2)=NC2=CC=CC=C2S(=O)(=O)C(C)C)C=C(C)C(=C1)C1CCNCC1 | 11.58/10.07 | 5.81 | 1 | 5.03 | ALK | */−/* | */−/* | −/*/* | + | +/− | [60] | ||
| Crizotinib | C[C@@H](OC1=CC(=CN=C1N)C1=CN(N=C1)C1CCNCC1)C1=C(Cl)C=CC(F)=C1Cl | Unknown/10.12 | 3.57 | 1 | 4.73 | ALK, ROS1 | */+/+ | + | − | [12,52] | ||||
| CT-707 | O=S(C1=C(NC2=C3C(NCC3)=NC(NC4=CC=C(N5CCC(N6CCN(C)CC6)CC5)C=C4OC)=N2)C=CC=C1)(NC(C)C)=O | 10.24/8.53 | 3.59 | 0 | ALK, FAK, PyK2 | |||||||||
| Dacomitinib | H]\C(CN1CCCCC1)=C(\[H])C(O)=NC1=C(OC)C=C2N=CN=C(NC3=CC(Cl)=C(F)C=C3)C2=C1 | −0.22/14.91 | 4.32 | 1 | 4.4 | EGFR | ||||||||
| Dasatinib | CC1=NC(NC2=NC=C(S2)C(=O)NC2=C(C)C=CC=C2Cl)=CC(=N1)N1CCN(CCO)CC1 | 8.49/7.22 | 3.82 | 1 | 2.24 | Bcr-Abl, Src | */−/+ | −/−/− | + | + | [12,52] | |||
| Defactinib | CNC(=O)C1=CC=C(NC2=NC=C(C(NCC3=C(N=CC=N3)N(C)S(C)(=O)=O)=N2)C(F)(F)F)C=C1 | 12.63/4.03 | 0.75 | 0 | −0.74 | FAK | ||||||||
| Dovitinib | CN1CCN(CC1)C1=CC=C2N=C(NC2=C1)C1=C(N)C2=C(NC1=O)C=CC=C2F | 8.56/7.87 | 1.35 | 1 | 1.59 | FGFR1/3, VEGFR, PDGFR, FLT3, c-KIT | − | − | [61] | |||||
| Ensartinib | ClC1=CC=C(F)C(Cl)=C1[C@@H](C)OC2=CC(C(NC3=CC=C(C(N4C[C@@H](C)N[C@@H](C)C4)=O)C=C3)=O)=NN=C2N | 13.82/8.00 | 3.95 | 0 | ALK | |||||||||
| Erlotinib | COCCOC1=CC2=C(C=C1OCCOC)C(NC1=CC(=CC=C1)C#C)=NC=N2 | 16–14/4.62 | 3.2 | 0 | 2.39 | EGFR | */−/+ | −/+/* | + | + | [12,52] | |||
| Famitinib | CCN(CC)CCN1CCC2=C(C(C)=C(N2)\C=C2/C(=O)NC3=CC=C(F)C=C23)C1=O | 11.46/9.01 | 2.68 | 1 | VEGFR | |||||||||
| Fruquintinib | CNC(=O)C1=C(C)OC2=CC(OC3=NC=NC4=CC(OC)=C(OC)C=C34)=CC=C12 | 14.99/2.57 | 2.64 | 0 | VEGFR | |||||||||
| Gefitinib | COC1=C(OCCCN2CCOCC2)C=C2C(NC3=CC(Cl)=C(F)C=C3)=NC=NC2=C1 | 16.11/6.85 | 3.75 | 0 | 4.11 | EGFR | */*/+ | −/−/* | + | + | [12,52] | |||
| Ibrutinib | NC1=NC=NC2=C1C(=NN2[C@@H]1CCCN(C1)C(=O)C=C)C1=CC=C(OC2=CC=CC=C2)C=C1 | 19.7/6.58 | 3.63 | 0 | 3.6 | BTK | ||||||||
| Icotinib | C#CC1=CC=CC(NC2=NC=NC3=CC4=C(OCCOCCOCCO4)C=C23)=C1 | Unknown/4.62 | 3.03 | 0 | EGFR | |||||||||
| Imatinib | CN1CCN(CC2=CC=C(C=C2)C(=O)NC2=CC(NC3=NC=CC(=N3)C3=CN=CC=C3)=C(C)C=C2)CC1 | 12.45/8.27 | 4.38 | 1 | 2.48 | Bcr-Abl | +/−/+ | −/−/− | +/−/− | */+ | + | + | +/* | [12,52] |
| Lapatinib | CS(=O)(=O)CCNCC1=CC=C(O1)C1=CC2=C(C=C1)N=CN=C2NC1=CC(Cl)=C(OCC2=CC(F)=CC=C2)C=C1 | 15.99/7.2 | 4.64 | 1 | 5.14 | HER2/neu, EGFR | + | + | [12,52] | |||||
| Lenvatinib | COC1=C(C=C2C(OC3=CC(Cl)=C(NC(O)=NC4CC4)C=C3)=CC=NC2=C1)C(O)=N | 3.56/6.1 | 2.16 | 1 | 2.8 | VEGFR | */−/− | −/*/− | −/−/* | + | + | [62] | ||
| Lorlatinib | C[C@H]1OC2=C(N)N=CC(=C2)C2=C(C#N)N(C)N=C2CN(C)C(=O)C2=C1C=C(F)C=C2 | 19.7/5.71 | 1.63 | 0 | ROS1, ALK | |||||||||
| Nilotinib | CC1=CN(C=N1)C1=CC(=CC(NC(=O)C2=CC(NC3=NC=CC(=N3)C3=CN=CC=C3)=C(C)C=C2)=C1)C(F)(F)F | 11.86/6.3 | 4.41 | 0 | 5.15 | Bcr-Abl | */+/− | −/−/− | + | + | [12,52] | |||
| Nintedanib | COC(=O)C1=CC=C2C(NC(=O)\C2=C(/NC2=CC=C(C=C2)N(C)C(=O)CN2CCN(C)CC2)C2=CC=CC=C2)=C1 | unknown/15.0 | 2.4 | 1 | 3.3 | VEGFR, FGFR, PDGFR | */−/− | */−/* | + | − | */− | [63] | ||
| Osimertinib | COC1=C(NC2=NC=CC(=N2)C2=CN(C)C3=C2C=CC=C3)C=C(NC(=O)C=C)C(=C1)N(C)CCN(C)C | 13.64/8.87 | 4.49 | 1 | 3.7 | EGFRT790M | */−/− | + | + | [64] | ||||
| Pazopanib | CN(C1=CC2=NN(C)C(C)=C2C=C1)C1=CC=NC(NC2=CC=C(C)C(=C2)S(N)(=O)=O)=N1 | 10.41/5.07 | 3.55 | 0 | 1.98 | VEGFR, FGFR, PDGFR, Kit, Itk, Lck, c-Fms | */+/+ | + | [12,52] | |||||
| Ponatinib | CN1CCN(CC2=CC=C(NC(=O)C3=CC(C#CC4=CN=C5C=CC=NN45)=C(C)C=C3)C=C2C(F)(F)F)CC1 | 11.36/8.03 | 4.97 | 1 | 3.1 | Bcr-Abl | */−/− | −/*/* | + | + | [12,52] | |||
| Poziotinib | COC1=C(OC2CCN(CC2)C(=O)C=C)C=C2C(NC3=CC=C(Cl)C(Cl)=C3F)=NC=NC2=C1 | 13.99/4.49 | 4.5 | 0 | EGFR, HER2/neu, Her 4 | |||||||||
| Pyrotinib | ClC1=CC(NC2=C(C#N)C=NC3=CC(OCC)=C(NC(/C=C/[C@@H]4N(C)CCC4)=O)C=C23)=CC=C1OCC5=CC=CC=N5 | 12.55/8.71 | 4.93 | 0 | HER2 | |||||||||
| Quizartinib | CC(C)(C)C1=CC(NC(=O)NC2=CC=C(C=C2)C2=CN3C(SC4=C3C=CC(OCCN3CCOCC3)=C4)=N2)=NO1 | 10.43/6.62 | 5.16 | 0 | FLT3 | |||||||||
| Regorafenib | CNC(=O)C1=CC(OC2=CC(F)=C(NC(=O)NC3=CC=C(Cl)C(=C3)C(F)(F)F)C=C2)=CC=N1 | 10.52/2.02 | 4.49 | 0 | 5.26 | VEGFR, TIE2, PDGFR-β, FGFR1, KIT, RET, c-RAF/RAF-1, BRAFV600E | */−/− | − | + | [12,52] | ||||
| Rociletinib | COC1=CC(=CC=C1NC1=NC=C(C(NC2=CC=CC(NC(=O)C=C)=C2)=N1)C(F)(F)F)N1CCN(CC1)C(C)=O | 13.63/3.62 | 4.41 | 0 | EGFRWT,T790M,L858R | |||||||||
| Ruxolitinib | N#CC[C@H](C1CCCC1)N1C=C(C=N1)C1=C2C=CNC2=NC=N1 | 13.89/5.51 | 5.51 | 0 | 2.1 | JAK1/2 | − | [65] | ||||||
| saracatinib | CN1CCN(CCOC2=CC3=C(C(NC4=C(Cl)C=CC5=C4OCO5)=NC=N3)C(OC3CCOCC3)=C2)CC1 | 11.81/8.19 | 3.53 | 1 | 2.74 | Bcr-Abl, Src | +/+/+ | +/− | [66] | |||||
| Savolitinib | C[C@H](N1N=NC2=NC=C(N=C12)C1=CN(C)N=C1)C1=CN2C=CN=C2C=C1 | Unknown/6.67 | 1.07 | 0 | c-Met | |||||||||
| Selumetinib | CN1C=NC2=C1C=C(C(O)=NOCCO)C(NC1=C(Cl)C=C(Br)C=C1)=C2F | 6.49/5.43 | 3.41 | 0 | 5.55 | MEK1/2 | + | + | [67] | |||||
| Semaxanib | CC1=CC(C)=C(N1)\C=C1/C(=O)NC2=CC=CC=C12 | 11.29/−2.1 | 2.98 | 0 | 2.87 | VEGFR2 | ||||||||
| Sorafenib | CNC(=O)C1=NC=CC(OC2=CC=C(NC(=O)NC3=CC(=C(Cl)C=C3)C(F)(F)F)C=C2)=C1 | 11.55/2.03 | 4.34 | 0 | 5.16 | VEGFR, PDGFR, Raf-1, C-Raf | −/+/− | */−/* | +/*/* | −/− | + | + | */+ | [12,48,52,68] |
| Sunitinib | CCN(CC)CCNC(=O)C1=C(C)NC(\C=C2/C(=O)NC3=C2C=C(F)C=C3)=C1C | 11.46/9.04 | 2.93 | 1 | 3.15 | VEGF, PDGF, c-KIT | −/+/− | + | + | [12,52] | ||||
| Tamatinib | COC1=CC(NC2=NC=C(F)C(NC3=NC4=C(OC(C)(C)C(=O)N4COP(O)(O)=O)C=C3)=N2)=CC(OC)=C1OC | 1.46/2.71 | 2.78 | 1 | SYK | |||||||||
| Tandutinib | COC1=C(OCCCN2CCCCC2)C=C2N=CN=C(N3CCN(CC3)C(=O)NC3=CC=C(OC(C)C)C=C3)C2=C1 | 14.01/9.03 | 4.34 | 1 | 4.38 | FLT3 | + | + | [69] | |||||
| Tivantinib | O=C1NC(=O)[C@H]([C@@H]1C1=CNC2=CC=CC=C12)C1=CN2CCCC3=C2C1=CC=C3 | 9.72/−8.6 | 3.27 | 0 | 3.26 | c-MET | ||||||||
| Trametinib | CN1C(=O)C(C)=C2N(C(=O)N(C3CC3)C(=O)C2=C1NC1=CC=C(I)C=C1F)C1=CC(NC(C)=O)=CC=C1 | 12.6/−3.7 | 3.18 | 0 | 2.68 | MEK | −/− | −/*/* | + | − | */− | [70] | ||
| Tofacitinib | [H][C@@]1(C)CCN(C[C@]1([H])N(C)C1=NC=NC2=C1C=CN2)C(=O)CC#N | 8.46/7.13 | 1.24 | 1 | 1.5 | JAK | */−/* | [71] | ||||||
| Vandetanib | COC1=C(OCC2CCN(C)CC2)C=C2N=CN=C(NC3=C(F)C=C(Br)C=C3)C2=C1 | 13.8/9.13 | 4.54 | 1 | 5.51 | VEGFR, EGFR, RET | + | − | [12,52] | |||||
| Vatalanib | ClC1=CC=C(NC2=NN=C(CC3=CC=NC=C3)C3=CC=CC=C23)C=C1 | 15.17/4.95 | 4.95 | 0 | 3.8 | VEGFR | ||||||||
| Vemurafenib | CCCS(=O)(=O)NC1=C(F)C(C(=O)C2=CNC3=NC=C(C=C23)C2=CC=C(Cl)C=C2)=C(F)C=C1 | 7.17/3.2 | 4.62 | 0 | 4.26 | BRAFV600E | + | [12,52] | ||||||
| Transporters | Genetic Polymorphism | Effects on TKIs | Reference |
|---|---|---|---|
| ABCB1 (P-gp) | 1199G > A | Increased efflux activity for: | [109] |
| Imatinib | |||
| Nilotinib | |||
| Dasatinib | |||
| 1236C > T-2677G > T-3435C > T (often inherited together in linkage disequilibrium) | TTT: Increased sensitivity to | [111] | |
| Imatinib and decreased efflux (compared to CGC wild type) | [12] | ||
| TTT: Increased resistance to sunitinib | |||
| CGT: Increased progression free survival | |||
| CGT: Increased toxicity for sorafenib | |||
| ABCG2 (BCRP) | 34G > A | G > A: decreased affinity and increased drug efficacy for imatinib, dasatinib, nilotinib, and bosutinib | [112] |
| GG: Decreased overall survival in NSCLC patients receiving EGFR-inhibitors (gefitinib, erlotinib and icotinib) compared to other genotypes | [113] | ||
| G > A: Increased major molecular response in imatinib | [12] | ||
| 421C > A | Reduced affinity and increased drug efficacy for imatinib, dasatinib, nilotinib, and bosutinib | [12,112] | |
| 623T > C | Loss of expression, increased drug efficacy for imatinib, dasatinib, nilotinib, and bosutinib | [112] | |
| 886G > C | Reduced affinity and increased drug efficacy for imatinib, dasatinib, nilotinib, and bosutinib | [112] | |
| 1574T > G | Reduced affinity and increased drug efficacy for imatinib, dasatinib, nilotinib, and bosutinib | [112] | |
| 1582G > A | increased drug efficacy for imatinib, dasatinib, and bosutinib | [112] | |
| SLC22A1 (OCT1) | 262Tdel | Truncated protein: reduced sensitivity for sorafenib | [68] |
| 181CGdelTins | Truncated protein: reduced sensitivity for sorafenib | [68] | |
| 1258ATGdel | Increased probability of treatment failure for imatinib | [12,118] | |
| 480G > C | Loss of sensitivity or treatment failure for imatinib | [12,126] | |
| SLC22A4 (OCTN1) | 1507C > T | TT genotype: Reduced major molecular response in imatinib | [12] |
| SLCO1B3 (OATP1B3) | 334T > G | TT genotype: Reduced complete molecular response in imatinib | [12] |
| Drug | Class | Tested in | References |
|---|---|---|---|
| Bafilomycin A1 | V-ATPase inhibitor | In vitro | [159] |
| Hydroxychloroquine/chloroquin | De-acidifier | clinical trials | [159] |
| Lys5 | De-acidifier | In vivo | [159] |
| Manzamine A | V-ATPase inhibitor | In vitro | [160,170] |
| Pifitrin-μ | HSP70 inhibitor | In vivo | [162] |
| Light sensitive drug | Photosensitizer | In vitro | [166] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Klerk, D.J.; Honeywell, R.J.; Jansen, G.; Peters, G.J. Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance. Cancers 2018, 10, 503. https://doi.org/10.3390/cancers10120503
De Klerk DJ, Honeywell RJ, Jansen G, Peters GJ. Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance. Cancers. 2018; 10(12):503. https://doi.org/10.3390/cancers10120503
Chicago/Turabian StyleDe Klerk, Daniel J., Richard J. Honeywell, Gerrit Jansen, and Godefridus J. Peters. 2018. "Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance" Cancers 10, no. 12: 503. https://doi.org/10.3390/cancers10120503
APA StyleDe Klerk, D. J., Honeywell, R. J., Jansen, G., & Peters, G. J. (2018). Transporter and Lysosomal Mediated (Multi)drug Resistance to Tyrosine Kinase Inhibitors and Potential Strategies to Overcome Resistance. Cancers, 10(12), 503. https://doi.org/10.3390/cancers10120503