AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
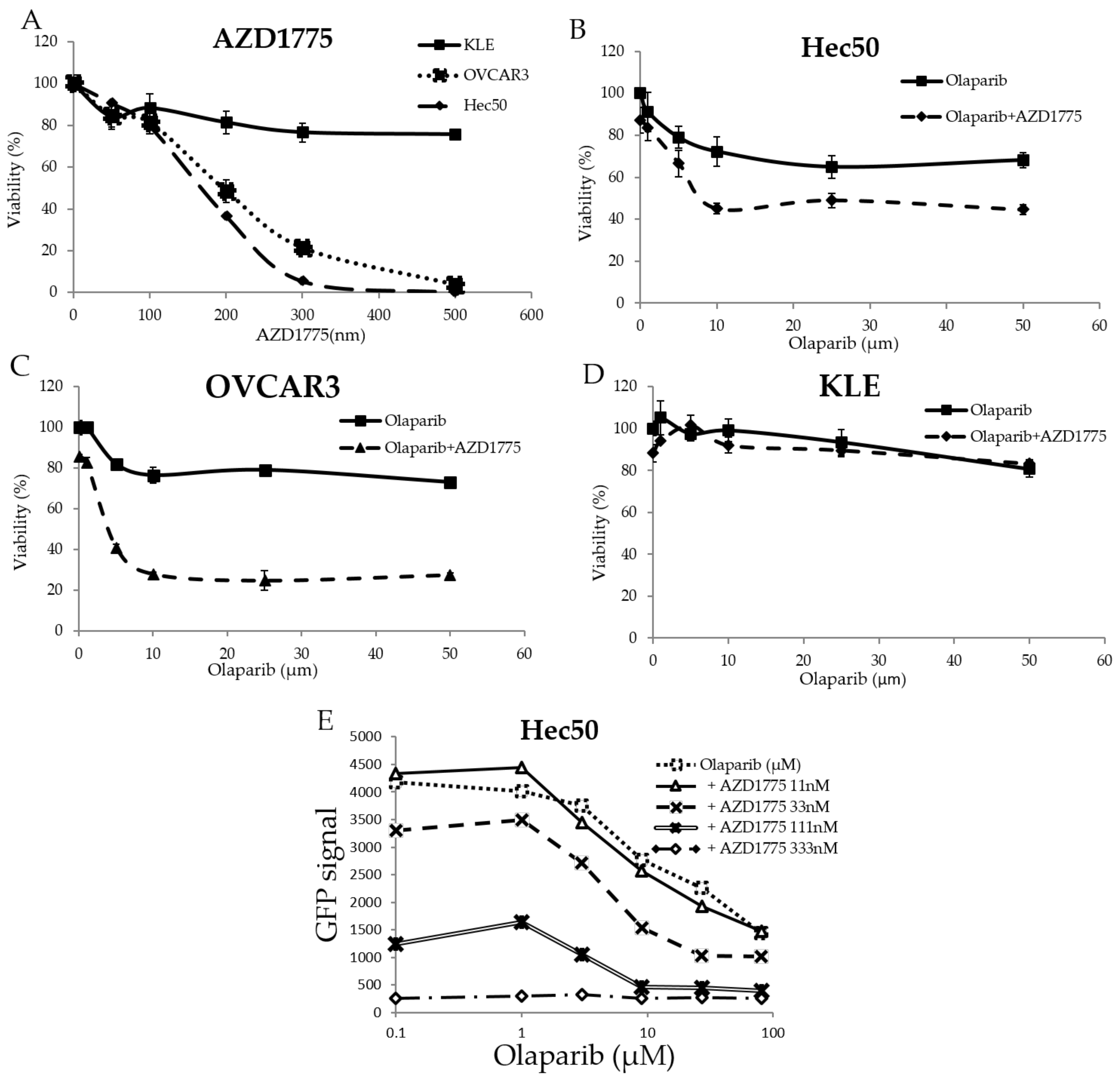
2.1. The WEE1 Inhibitor AZD1775 Increases Sensitivity to the PARP Inhibitor Olaparib
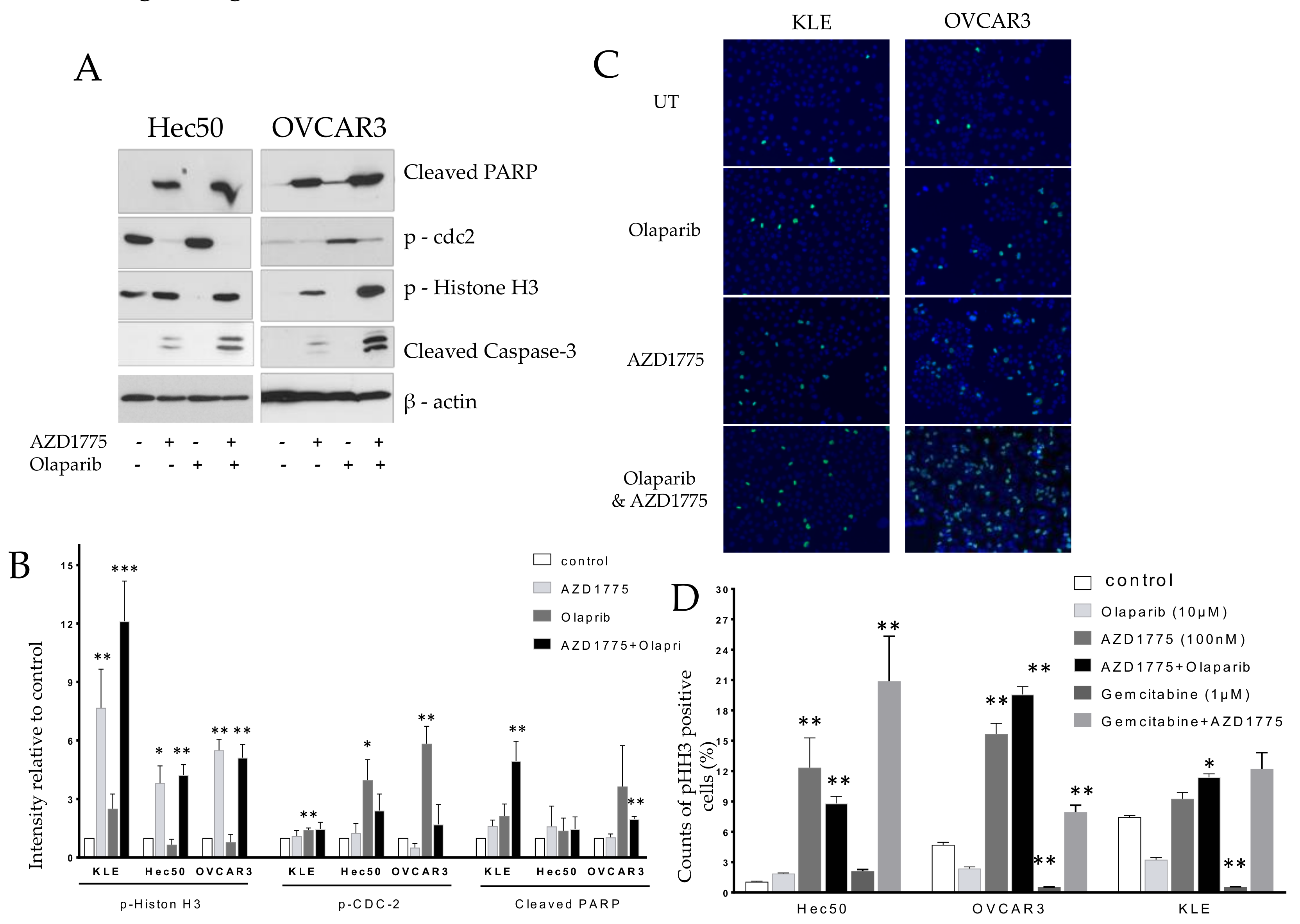
2.2. Olaparib Combined with AZD1775 Increases Cell Death
2.3. AZD1775 Increases Sensitivity to Gemcitabine
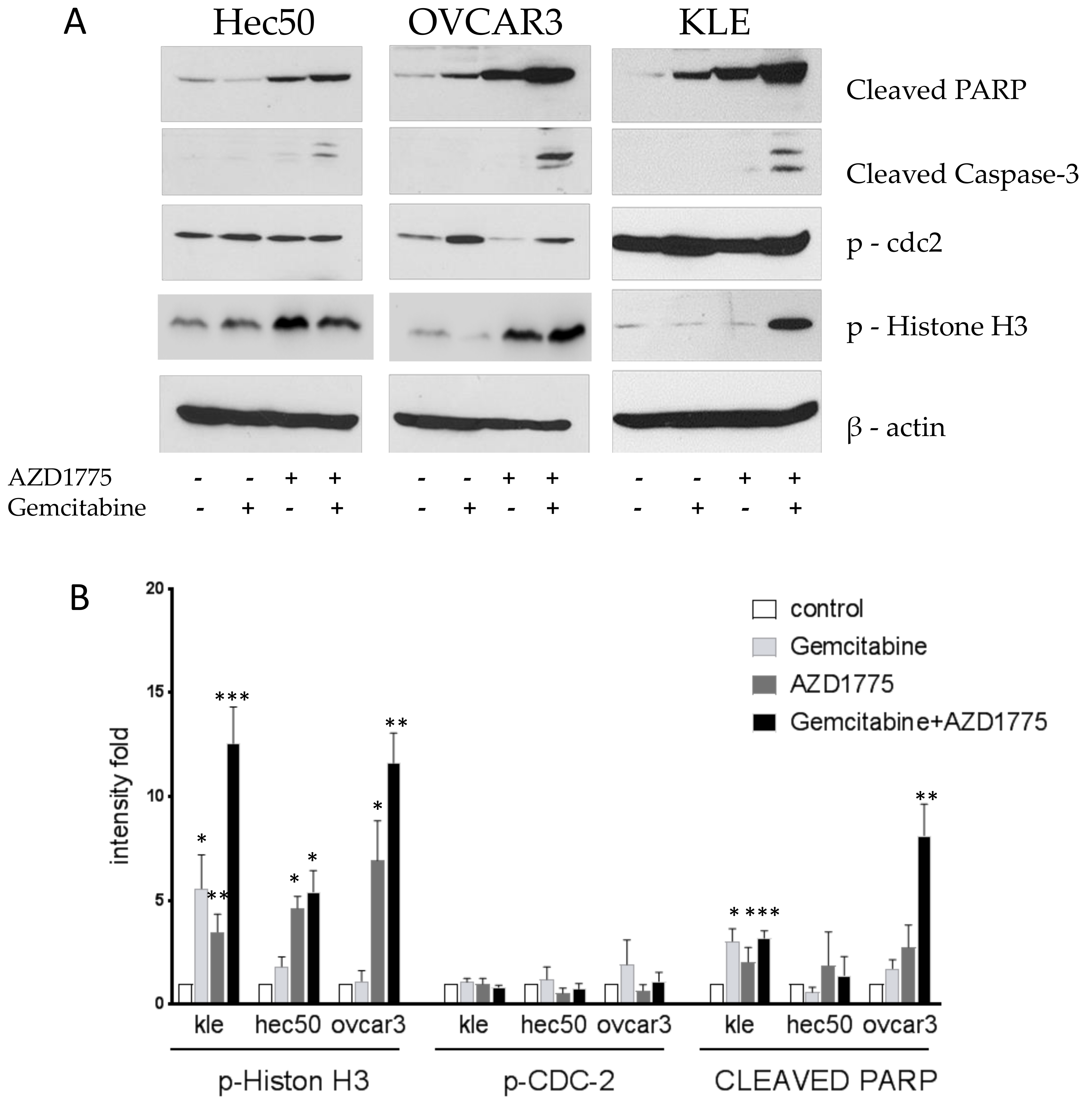
2.4. Gemcitabine and AZD1775 Combination Increases Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drug Treatment and Cell Viability Assays
4.3. Western Blotting
4.4. Antibodies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Yao, S.L.; Akhtar, A.J.; McKenna, K.A.; Bedi, G.C.; Sidransky, D.; Mabry, M.; Ravi, R.; Collector, M.I.; Jones, R.J.; Sharkis, S.J.; et al. Selective radiosensitization of p53-deficient cells by caffeine-mediated activation of p34cdc2 kinase. Nat. Med. 1996, 2, 1140–1143. [Google Scholar] [CrossRef] [PubMed]
- Brachova, P.; Thiel, K.W.; Leslie, K.K. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 19257–19275. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Beijnen, J.H.; Schellens, J.H. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr. Clin. Pharmacol. 2010, 5, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Yamanaka, K.; Itadani, H.; Arai, T.; Nishibata, T.; Hirai, H.; Kotani, H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug WEE1 inhibitor. Mol. Cancer 2009, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule WEE1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Doroshow, J.H.; Kummar, S. WEE1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3159–3164. [Google Scholar] [CrossRef] [PubMed]
- Garcia, T.B.; Snedeker, J.C.; Baturin, D.; Gardner, L.; Fosmire, S.P.; Zhou, C.; Jordan, C.T.; Venkataraman, S.; Vibhakar, R.; Porter, C.C. A Small-Molecule Inhibitor of WEE1, AZD1775, Synergizes with Olaparib by Impairing Homologous Recombination and Enhancing DNA Damage and Apoptosis in Acute Leukemia. Mol. Cancer Ther. 2017, 16, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, N.V.; De Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; Demuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive, relapsed serous ovarian cancer and a BRCA mutation: Overall survival adjusted for postprogression poly(adenosine diphosphate ribose) polymerase inhibitor therapy. Cancer 2016, 122, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.; Hansen, O.P.; Theilade, K.; Hansen, M.; Neijt, J.P. Phase II study of gemcitabine (2′,2′-difluorodeoxycytidine) in previously treated ovarian cancer patients. J. Natl. Cancer Inst. 1994, 86, 1530–1533. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [Google Scholar] [PubMed]
- Xie, G.; Habbersett, R.C.; Jia, Y.; Peterson, S.R.; Lehnert, B.E.; Bradbury, E.M.; D’Anna, J.A. Requirements for p53 and the ATM gene product in the regulation of G1/S and S phase checkpoints. Oncogene 1998, 16, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Webster, P.J.; Littlejohns, A.T.; Gaunt, H.J.; Prasad, K.R.; Beech, D.J.; Burke, D.A. AZD1775 induces toxicity through double-stranded DNA breaks independently of chemotherapeutic agents in p53-mutated colorectal cancer cells. Cell Cycle 2017, 16, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kulesskiy, E.; Saarela, J.; Turunen, L.; Wennerberg, K.; Aittokallio, T.; Tang, J. Methods for High-throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 2018, 1711, 351–398. [Google Scholar] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, X.; Bi, J.; Li, Y.; Yang, S.; Zhang, Y.; Li, M.; Liu, H.; Li, Y.; Mcdonald, M.E.; Thiel, K.W.; et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers 2018, 10, 149. https://doi.org/10.3390/cancers10050149
Meng X, Bi J, Li Y, Yang S, Zhang Y, Li M, Liu H, Li Y, Mcdonald ME, Thiel KW, et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers. 2018; 10(5):149. https://doi.org/10.3390/cancers10050149
Chicago/Turabian StyleMeng, Xiangbing, Jianling Bi, Yujun Li, Shujie Yang, Yuping Zhang, Mary Li, Haitao Liu, Yiyang Li, Megan E. Mcdonald, Kristina W. Thiel, and et al. 2018. "AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations" Cancers 10, no. 5: 149. https://doi.org/10.3390/cancers10050149
APA StyleMeng, X., Bi, J., Li, Y., Yang, S., Zhang, Y., Li, M., Liu, H., Li, Y., Mcdonald, M. E., Thiel, K. W., Wen, K.-K., Wang, X., Wu, M., & Leslie, K. K. (2018). AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers, 10(5), 149. https://doi.org/10.3390/cancers10050149