Exploiting NK Cell Surveillance Pathways for Cancer Therapy



{kind=link}
Abstract
:1. Introduction
2. Killer Cell Ig-Like Receptors (KIR)
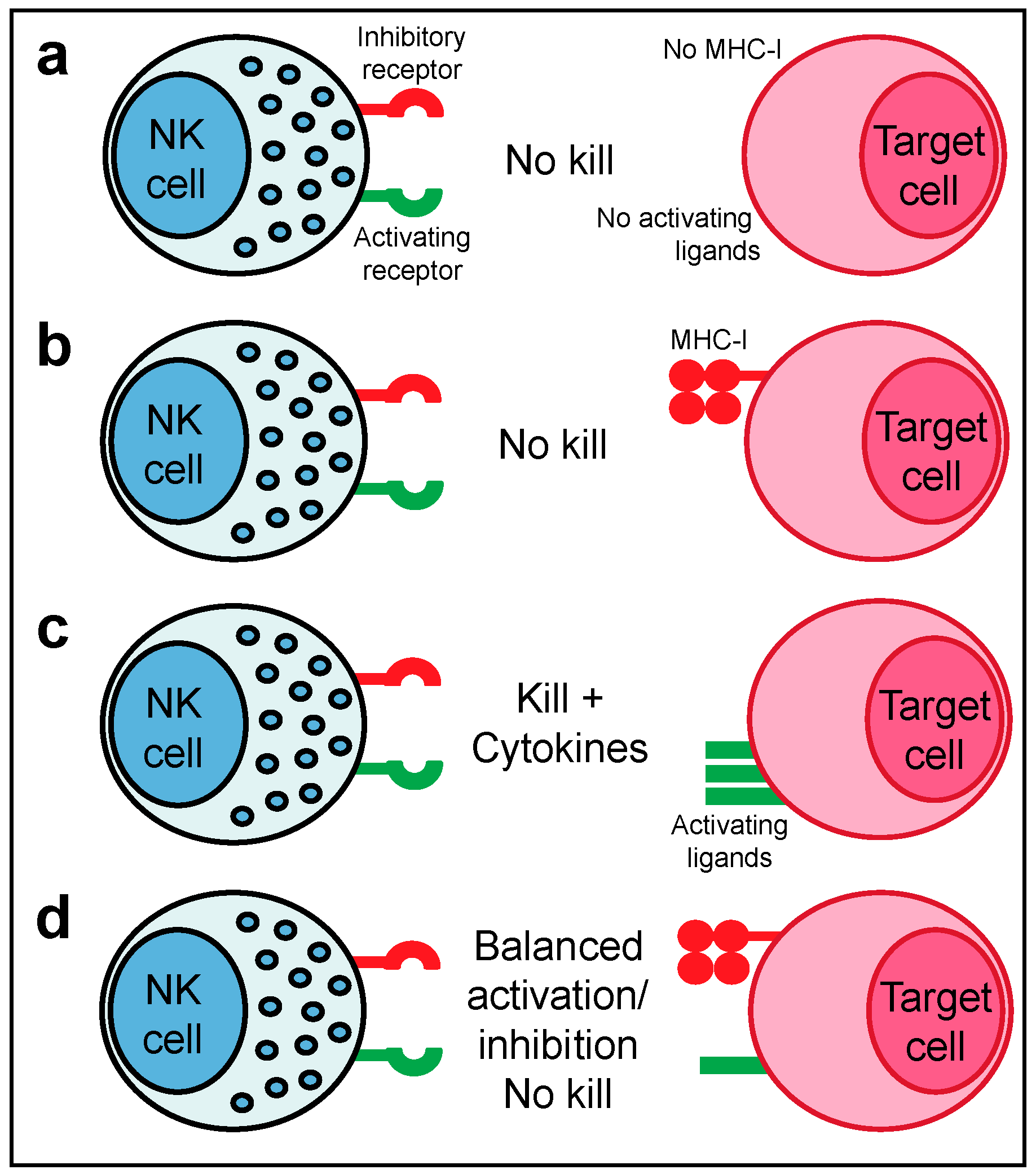
2.1. NK Cell Education
2.2. KIR and Haematopoietic Stem Cell Transplantation (HCST)
3. Monoclonal Antibodies for Cancer Immunotherapy
3.1. PD-1
3.2. NKG2A
3.3. T-Cell Immunoglobulin and Mucin-Domain-Containing-3 (Tim-3)
3.4. T-Cell Immunoreceptor with Immunoglobulin and Immunoreceptor Tyrosine-Based Inhibition Motif Domains (TIGIT)
3.5. Interleukin-1 Receptor 8 (IL-1R8)
3.6. Sialic Acid Binding Immunoglobulin-Like Lectins (Siglecs)
4. Augmenting Activating NK Cell Receptor Pathways
4.1. CD16
4.2. Signalling Lymphocytic Activation Molecules Family 7 (SLAMF7)
4.3. Natural Killer Group 2D (NKG2D)
5. Recombinant Approaches to Cancer Immunotherapy
5.1. NKG2D Chimeric Antigen Receptors (CARs)
5.2. Bi- and Tri-Specific Killer Engagers (BiKEs and TriKEs)
6. Chemotherapy
7. Conclusions
Funding
Conflicts of Interest
References
- Müller-Hermelink, N.; Braumüller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A.; et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Aquino-López, A.; Senyukov, V.V.; Vlasic, Z.; Kleinerman, E.S.; Lee, D.A. Interferon Gamma Induces Changes in Natural Killer (NK) Cell Ligand Expression and Alters NK Cell-Mediated Lysis of Pediatric Cancer Cell Lines. Front. Immunol. 2017, 8, 391. [Google Scholar] [CrossRef]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534.e19–548.e19. [Google Scholar] [CrossRef]
- Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef]
- Su, X.; Yu, Y.; Zhong, Y.; Giannopoulou, E.G.; Hu, X.; Liu, H.; Cross, J.R.; Rätsch, G.; Rice, C.M.; Ivashkiv, L.B. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015, 16, 838–849. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Semeraro, M.; Sarabi, M.; Desbois, M.; Locher, C.; Mendez, R.; Vimond, N.; Concha, A.; Garrido, F.; Isambert, N.; et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. 2013, 73, 3499–3510. [Google Scholar] [CrossRef]
- Mitchell, G.; Isberg, R.R. Innate Immunity to Intracellular Pathogens: Balancing Microbial Elimination and Inflammation. Cell Host Microbe 2017, 22, 166–175. [Google Scholar] [CrossRef]
- Pierini, R.; Perret, M.; Djebali, S.; Juruj, C.; Michallet, M.-C.; Förster, I.; Marvel, J.; Walzer, T.; Henry, T. ASC controls IFN-γ levels in an IL-18-dependent manner in caspase-1-deficient mice infected with Francisella novicida. J. Immunol. 2013, 191, 3847–3857. [Google Scholar] [CrossRef]
- Li, S.S.; Ogbomo, H.; Mansour, M.K.; Xiang, R.F.; Szabo, L.; Munro, F.; Mukherjee, P.; Mariuzza, R.A.; Amrein, M.; Vyas, J.M.; et al. Identification of the fungal ligand triggering cytotoxic PRR-mediated NK cell killing of Cryptococcus and Candida. Nat. Commun. 2018, 9, 751. [Google Scholar] [CrossRef]
- Raulet, D.H.; Marcus, A.; Coscoy, L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev. 2017, 280, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Ljunggren, H.G.; Kärre, K. In search of the “missing self”: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Kadri, N.; Thanh, T.L.; Höglund, P. Selection, tuning, and adaptation in mouse NK cell education. Immunol. Rev. 2015, 267, 167–177. [Google Scholar] [CrossRef]
- Bix, M.; Liao, N.S.; Zijlstra, M.; Loring, J.; Jaenisch, R.; Raulet, D. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature 1991, 349, 329–331. [Google Scholar] [CrossRef]
- Liao, N.S.; Bix, M.; Zijlstra, M.; Jaenisch, R.; Raulet, D. MHC class I deficiency: Susceptibility to natural killer (NK) cells and impaired NK activity. Science 1991, 253, 199–202. [Google Scholar] [CrossRef]
- Höglund, P.; Ohlén, C.; Carbone, E.; Franksson, L.; Ljunggren, H.G.; Latour, A.; Koller, B.; Kärre, K. Recognition of beta 2-microglobulin-negative (beta 2m-) T-cell blasts by natural killer cells from normal but not from beta 2m-mice: Nonresponsiveness controlled by beta 2m- bone marrow in chimeric mice. Proc. Natl. Acad. Sci. USA 1991, 88, 10332–10336. [Google Scholar] [CrossRef]
- Parham, P.; Guethlein, L.A. Genetics of Natural Killer Cells in Human Health, Disease, and Survival. Annu. Rev. Immunol. 2018, 36, 519–548. [Google Scholar] [CrossRef]
- Anfossi, N.; André, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Guethlein, L.A.; Norman, P.J.; Hilton, H.G.; Parham, P. Co-evolution of MHC class I and variable NK cell receptors in placental mammals. Immunol. Rev. 2015, 267, 259–282. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Brooks, E.G.; Falco, M.; Ferrara, G.B.; Strominger, J.L. Generation of allospecific natural killer cells by stimulation across a polymorphism of HLAC. Science. 1993, 260, 1121–1124. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Miller, J.S.; Cooley, S.; Parham, P.; Farag, S.S.; Verneris, M.R.; McQueen, K.L.; Guethlein, L.A.; Trachtenberg, E.A.; Haagenson, M.; Horowitz, M.M.; et al. Missing KIR ligands are associated with less relapse and increased graft-versus-host disease (GVHD) following unrelated donor allogeneic HCT. Blood 2007, 109, 5058–5061. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Mancusi, A.; Capanni, M.; Urbani, E.; Carotti, A.; Aloisi, T.; Stern, M.; Pende, D.; Perruccio, K.; Burchielli, E.; et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: Challenging its predictive value. Blood 2007, 110, 433–440. [Google Scholar] [CrossRef]
- Hsu, K.C.; Keever-Taylor, C.A.; Wilton, A.; Pinto, C.; Heller, G.; Arkun, K.; O’Reilly, R.J.; Horowitz, M.M.; Dupont, B. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood 2005, 105, 4878–4884. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Venstrom, J.M.; Liu, X.-R.; Pring, J.; Hasan, R.S.; O’Reilly, R.J.; Hsu, K.C. Breaking tolerance to self, circulating natural killer cells expressing inhibitory KIR for non-self HLA exhibit effector function after T cell–depleted allogeneic hematopoietic cell transplantation. Blood 2009, 113, 3875–3884. [Google Scholar] [CrossRef]
- Boudreau, J.E.; Hsu, K.C. Natural Killer Cell Education and the Response to Infection and Cancer Therapy: Stay Tuned. Trends Immunol. 2018, 39, 222–239. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Pitt, M.; Curtsinger, J.; Luo, X.; Lopez-Vergès, S.; Lanier, L.L.; Weisdorf, D.; Miller, J.S. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012, 119, 2665–2674. [Google Scholar] [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Schwartz, B.S.; Pando, M.J.; Jarjoura, J.; York, V.A.; Houchins, J.P.; Miller, S.; Kang, S.-M.; Norris, P.J.; et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 14725–14732. [Google Scholar] [CrossRef]
- Cichocki, F.; Cooley, S.; DeFor, T.E.; Schlums, H.; Zhang, B.; Brunstein, C.G.; Blazar, B.R.; Wagner, J.; Diamond, D.J.; Verneris, M.R.; et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016, 30, 456–463. [Google Scholar] [CrossRef]
- Tarek, N.; Le Luduec, J.-B.; Gallagher, M.M.; Zheng, J.; Venstrom, J.M.; Chamberlain, E.; Modak, S.; Heller, G.; Dupont, B.; Cheung, N.-K.V.; et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J. Clin. Investig. 2012, 122, 3260–3270. [Google Scholar] [CrossRef] [Green Version]
- Erbe, A.K.; Wang, W.; Reville, P.K.; Carmichael, L.; Kim, K.; Mendonca, E.A.; Song, Y.; Hank, J.A.; London, W.B.; Naranjo, A.; et al. HLA-Bw4-I-80 Isoform Differentially Influences Clinical Outcome as Compared to HLA-Bw4-T-80 and HLA-A-Bw4 Isoforms in Rituximab or Dinutuximab-Based Cancer Immunotherapy. Front. Immunol. 2017, 8, 675. [Google Scholar] [CrossRef]
- Erbe, A.K.; Wang, W.; Carmichael, L.; Kim, K.; Mendonça, E.A.; Song, Y.; Hess, D.; Reville, P.K.; London, W.B.; Naranjo, A.; et al. Neuroblastoma Patients’ KIR and KIR-Ligand Genotypes Influence Clinical Outcome for Dinutuximab-based Immunotherapy: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2018, 24, 189–196. [Google Scholar] [CrossRef]
- Forlenza, C.J.; Boudreau, J.E.; Zheng, J.; Le Luduec, J.-B.; Chamberlain, E.; Heller, G.; Cheung, N.-K.V.; Hsu, K.C. KIR3DL1 Allelic Polymorphism and HLA-B Epitopes Modulate Response to Anti-GD2 Monoclonal Antibody in Patients with Neuroblastoma. J. Clin. Oncol. 2016, 34, 2443–2451. [Google Scholar] [CrossRef]
- Boudreau, J.E.; Giglio, F.; Gooley, T.A.; Stevenson, P.A.; Le Luduec, J.-B.; Shaffer, B.C.; Rajalingam, R.; Hou, L.; Hurley, C.K.; Noreen, H.; et al. KIR3DL1/ HL A-B Subtypes Govern Acute Myelogenous Leukemia Relapse After Hematopoietic Cell Transplantation. J. Clin. Oncol. 2017, 35, 2268–2278. [Google Scholar] [CrossRef]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Baars, P.A.; Ribeiro Do Couto, L.M.; Leusen, J.H.; Hooibrink, B.; Kuijpers, T.W.; Lens, S.M.; van Lier, R.A. Cytolytic mechanisms and expression of activation-regulating receptors on effector-type CD8+CD45RA+CD27- human T cells. J. Immunol. 2000, 165, 1910–1917. [Google Scholar] [CrossRef]
- Odorizzi, P.M.; Wherry, E.J. Inhibitory receptors on lymphocytes: Insights from infections. J. Immunol. 2012, 188, 2957–2965. [Google Scholar] [CrossRef]
- Rygiel, T.P.; Rijkers, E.S.K.; de Ruiter, T.; Stolte, E.H.; van der Valk, M.; Rimmelzwaan, G.F.; Boon, L.; van Loon, A.M.; Coenjaerts, F.E.; Hoek, R.M.; et al. Lack of CD200 enhances pathological T cell responses during influenza infection. J. Immunol. 2009, 183, 1990–1996. [Google Scholar] [CrossRef]
- Frebel, H.; Nindl, V.; Schuepbach, R.A.; Braunschweiler, T.; Richter, K.; Vogel, J.; Wagner, C.A.; Loffing-Cueni, D.; Kurrer, M.; Ludewig, B.; et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J. Exp. Med. 2012, 209, 2485–2499. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Waitz, R.; Fassò, M.; Allison, J.P. CTLA-4 blockade synergizes with cryoablation to mediate tumor rejection. Oncoimmunology 2012, 1, 544–546. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.K.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef]
- Munir, S.; Andersen, G.H.; Svane, I.M.; Andersen, M.H. The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology 2013, 2, e23991. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335.e3–346.e3. [Google Scholar] [CrossRef]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of PD-L1 on tumor cells is associated with reduced MHC I expression and PD-L1 expression increases in recurrent serous ovarian cancer. Sci. Rep. 2017, 7, 42929. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259.e11–1271.e11. [Google Scholar] [CrossRef]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272.e15–1283.e15. [Google Scholar] [CrossRef]
- Roemer, M.G.M.; Advani, R.H.; Redd, R.A.; Pinkus, G.S.; Natkunam, Y.; Ligon, A.H.; Connelly, C.F.; Pak, C.J.; Carey, C.D.; Daadi, S.E.; et al. Classical Hodgkin Lymphoma with Reduced β2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol. Res. 2016, 4, 910–916. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.-C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Vance, R.E.; Kraft, J.R.; Altman, J.D.; Jensen, P.E.; Raulet, D.H. Mouse CD94/NKG2A is a natural killer cell receptor for the nonclassical major histocompatibility complex (MHC) class I molecule Qa-1(b). J. Exp. Med. 1998, 188, 1841–1848. [Google Scholar] [CrossRef]
- Hammer, Q.; Rückert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef]
- Zeng, L.; Sullivan, L.C.; Vivian, J.P.; Walpole, N.G.; Harpur, C.M.; Rossjohn, J.; Clements, C.S.; Brooks, A.G. A structural basis for antigen presentation by the MHC class Ib molecule, Qa-1b. J. Immunol. 2012, 188, 302–310. [Google Scholar] [CrossRef]
- Rapaport, A.S.; Schriewer, J.; Gilfillan, S.; Hembrador, E.; Crump, R.; Plougastel, B.F.; Wang, Y.; Le Friec, G.; Gao, J.; Cella, M.; et al. The Inhibitory Receptor NKG2A Sustains Virus-Specific CD8+ T Cells in Response to a Lethal Poxvirus Infection. Immunity 2015, 43, 1112–1124. [Google Scholar] [CrossRef]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744.e15–1755.e15. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731.e13–1743.e13. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef]
- Rangachari, M.; Zhu, C.; Sakuishi, K.; Xiao, S.; Karman, J.; Chen, A.; Angin, M.; Wakeham, A.; Greenfield, E.A.; Sobel, R.A.; et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat. Med. 2012, 18, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Leitner, J.; Rieger, A.; Pickl, W.F.; Zlabinger, G.; Grabmeier-Pfistershammer, K.; Steinberger, P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. 2013, 9, e1003253. [Google Scholar] [CrossRef]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef]
- Avery, L.; Filderman, J.; Szymczak-Workman, A.L.; Kane, L.P. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc. Natl. Acad. Sci. USA 2018, 115, 2455–2460. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [Green Version]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Gallois, A.; Silva, I.; Osman, I.; Bhardwaj, N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Oncoimmunology 2014, 3, e946365. [Google Scholar] [CrossRef]
- Anderson, A.C.; Anderson, D.E.; Bregoli, L.; Hastings, W.D.; Kassam, N.; Lei, C.; Chandwaskar, R.; Karman, J.; Su, E.W.; Hirashima, M.; et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 2007, 318, 1141–1143. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef]
- Minnie, S.A.; Kuns, R.D.; Gartlan, K.H.; Zhang, P.; Wilkinson, A.N.; Samson, L.; Guillerey, C.; Engwerda, C.; MacDonald, K.P.A.; Smyth, M.J.; et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018, 132, 1675–1688. [Google Scholar] [CrossRef]
- Garlanda, C.; Riva, F.; Polentarutti, N.; Buracchi, C.; Sironi, M.; De Bortoli, M.; Muzio, M.; Bergottini, R.; Scanziani, E.; Vecchi, A.; et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA 2004, 101, 3522–3526. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Qian, Y.; Yao, J.; Grace, C.; Li, X. SIGIRR inhibits interleukin-1 receptor- and toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem. 2005, 280, 25233–25241. [Google Scholar] [CrossRef]
- Bulek, K.; Swaidani, S.; Qin, J.; Lu, Y.; Gulen, M.F.; Herjan, T.; Min, B.; Kastelein, R.A.; Aronica, M.; Kosz-Vnenchak, M.; et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J. Immunol. 2009, 182, 2601–2609. [Google Scholar] [CrossRef]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef]
- Bertilaccio, M.T.S.; Simonetti, G.; Dagklis, A.; Rocchi, M.; Rodriguez, T.V.; Apollonio, B.; Mantovani, A.; Ponzoni, M.; Ghia, P.; Garlanda, C.; et al. Lack of TIR8/SIGIRR triggers progression of chronic lymphocytic leukemia in mouse models. Blood 2011, 118, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popović, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef] [Green Version]
- El-Darawish, Y.; Li, W.; Yamanishi, K.; Pencheva, M.; Oka, N.; Yamanishi, H.; Matsuyama, T.; Tanaka, Y.; Minato, N.; Okamura, H. Frontline Science: IL-18 primes murine NK cells for proliferation by promoting protein synthesis, survival, and autophagy. J. Leukoc. Biol. 2018, 104, 253–264. [Google Scholar] [CrossRef]
- Hammer, Q.; Rückert, T.; Dunst, J.; Romagnani, C. Adaptive Natural Killer Cells Integrate Interleukin-18 during Target-Cell Encounter. Front. Immunol. 2017, 8, 1976. [Google Scholar] [CrossRef]
- Varki, A.; Gagneux, P. Biological Functions of Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Cao, H.; de Bono, B.; Belov, K.; Wong, E.S.; Trowsdale, J.; Barrow, A.D. Comparative genomics indicates the mammalian CD33rSiglec locus evolved by an ancient large-scale inverse duplication and suggests all Siglecs share a common ancestral region. Immunogenetics 2009, 61, 401–417. [Google Scholar] [CrossRef]
- Cao, H.; Lakner, U.; de Bono, B.; Traherne, J.A.; Trowsdale, J.; Barrow, A.D. SIGLEC16 encodes a DAP12-associated receptor expressed in macrophages that evolved from its inhibitory counterpart SIGLEC11 and has functional and non-functional alleles in humans. Eur. J. Immunol. 2008, 38, 2303–2315. [Google Scholar] [CrossRef]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation Changes in Cancer. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Belisle, J.A.; Horibata, S.; Jennifer, G.A.A.; Petrie, S.; Kapur, A.; André, S.; Gabius, H.-J.; Rancourt, C.; Connor, J.; Paulson, J.C.; et al. Identification of Siglec-9 as the receptor for MUC16 on human NK cells, B cells, and monocytes. Mol. Cancer 2010, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Jandus, C.; Boligan, K.F.; Chijioke, O.; Liu, H.; Dahlhaus, M.; Démoulins, T.; Schneider, C.; Wehrli, M.; Hunger, R.E.; Baerlocher, G.M.; et al. Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Investig. 2014, 124, 1810–1820. [Google Scholar] [CrossRef]
- Shao, J.-Y.; Yin, W.-W.; Zhang, Q.-F.; Liu, Q.; Peng, M.-L.; Hu, H.-D.; Hu, P.; Ren, H.; Zhang, D.-Z. Siglec-7 Defines a Highly Functional Natural Killer Cell Subset and Inhibits Cell-Mediated Activities. Scand. J. Immunol. 2016, 84, 182–190. [Google Scholar] [CrossRef]
- Ogata, S.; Maimonis, P.J.; Itzkowitz, S.H. Mucins bearing the cancer-associated sialosyl-Tn antigen mediate inhibition of natural killer cell cytotoxicity. Cancer Res. 1992, 52, 4741–4746. [Google Scholar]
- Van Rinsum, J.; Smets, L.A.; Van Rooy, H.; Van den Eijnden, D.H. Specific inhibition of human natural killer cell-mediated cytotoxicity by sialic acid and sialo-oligosaccharides. Int. J. Cancer 1986, 38, 915–922. [Google Scholar] [CrossRef]
- Yogeeswaran, G.; Gronberg, A.; Hansson, M.; Dalianis, T.; Kiessling, R.; Welsh, R.M. Correlation of glycosphingolipids and sialic acid in YAC-1 lymphoma variants with their sensitivity to natural killer-cell-mediated lysis. Int. J. Cancer 1981, 28, 517–526. [Google Scholar] [CrossRef]
- Stanczak, M.A.; Siddiqui, S.S.; Trefny, M.P.; Thommen, D.S.; Boligan, K.F.; Gunten, S.; von Tzankov, A.; Tietze, L.; Lardinois, D.; Heinzelmann-Schwarz, V.; et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Investig. 2018, 128, 4912–4923. [Google Scholar] [CrossRef]
- Schwarz, F.; Landig, C.S.; Siddiqui, S.; Secundino, I.; Olson, J.; Varki, N.; Nizet, V.; Varki, A. Paired Siglec receptors generate opposite inflammatory responses to a human-specific pathogen. EMBO J. 2017, 36, 751–760. [Google Scholar] [CrossRef]
- Lanier, L.L.; Ruitenberg, J.J.; Phillips, J.H. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. J. Immunol. 1988, 141, 3478–3485. [Google Scholar]
- Vivier, E.; da Silva, A.J.; Ackerly, M.; Levine, H.; Rudd, C.E.; Anderson, P. Association of a 70-kDa tyrosine phosphoprotein with the CD16: Zeta: Gamma complex expressed in human natural killer cells. Eur. J. Immunol. 1993, 23, 1872–1876. [Google Scholar] [CrossRef]
- Vivier, E.; Ackerly, M.; Rochet, N.; Anderson, P. Structure and function of the CD16:Zeta:Gamma complex expressed on human natural-killer cells. Int. J. Cancer Suppl. 1992, 7, 11–14. [Google Scholar]
- Congy-Jolivet, N.; Bolzec, A.; Ternant, D.; Ohresser, M.; Watier, H.; Thibault, G. Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. 2008, 68, 976–980. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.-G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Gil-Krzewska, A.; Peruzzi, G.; Borrego, F. Matrix metalloproteinases inhibition promotes the polyfunctionality of human natural killer cells in therapeutic antibody-based anti-tumour immunotherapy. Clin. Exp. Immunol. 2013, 173, 131–139. [Google Scholar] [CrossRef]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.K.; Miller, J.S. Current strategies exploiting NK-cell therapy to treat haematologic malignancies. Int. J. Immunogenet. 2018. [Google Scholar] [CrossRef]
- Wu, N.; Veillette, A. SLAM family receptors in normal immunity and immune pathologies. Curr. Opin. Immunol. 2016, 38, 45–51. [Google Scholar] [CrossRef]
- Tassi, I.; Colonna, M. The cytotoxicity receptor CRACC (CS-1) recruits EAT-2 and activates the PI3K and phospholipase Cgamma signaling pathways in human NK cells. J. Immunol. 2005, 175, 7996–8002. [Google Scholar] [CrossRef]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.-F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef] [Green Version]
- Friend, R.; Bhutani, M.; Voorhees, P.M.; Usmani, S.Z. Clinical potential of SLAMF7 antibodies—Focus on elotuzumab in multiple myeloma. Drug Des. Dev. Ther. 2017, 11, 893–900. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Bacon, L.; Eagle, R.A.; Meyer, M.; Easom, N.; Young, N.T.; Trowsdale, J. Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J. Immunol. 2004, 173, 1078–1084. [Google Scholar] [CrossRef]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Cao, W.; Xi, X.; Wang, Z.; Dong, L.; Hao, Z.; Cui, L.; Ma, C.; He, W. Four novel ULBP splice variants are ligands for human NKG2D. Int. Immunol. 2008, 20, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Eagle, R.A.; Flack, G.; Warford, A.; Martínez-Borra, J.; Jafferji, I.; Traherne, J.A.; Ohashi, M.; Boyle, L.H.; Barrow, A.D.; Caillat-Zucman, S.; et al. Cellular expression, trafficking, and function of two isoforms of human ULBP5/RAET1G. PLoS ONE 2009, 4, e4503. [Google Scholar] [CrossRef]
- Lanier, L.L. NKG2D receptor and its ligands in host defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Jafferji, I.; Barrow, A.D. Beyond Stressed Self: Evidence for NKG2D Ligand Expression on Healthy Cells. Curr. Immunol. Rev. 2009, 5, 22–34. [Google Scholar] [CrossRef]
- Dhar, P.; Wu, J.D. NKG2D and its ligands in cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Brayer, J.; Sagatys, E.M.; Lonez, C.; Breman, E.; Agaugué, S.; Verma, B.; Gilham, D.E.; Lehmann, F.F.; Davila, M.L. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica 2018, 103, e424–e426. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Pan, J.; Du, C.; Si, W.; Yao, M.; Xu, L.; Zheng, H.; Xu, M.; Chen, D.; Wang, S.; et al. Silencing NKG2D ligand-targeting miRNAs enhances natural killer cell-mediated cytotoxicity in breast cancer. Cell Death Dis. 2017, 8, e2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, S.; Kretzmer, L.Z.; Koues, O.I.; Payton, J.E.; Oltz, E.M.; Cashen, A.; Polic, B.; Schreiber, R.D.; Shaw, A.S.; Markiewicz, M.A. NKG2D-NKG2D Ligand Interaction Inhibits the Outgrowth of Naturally Arising Low-Grade B Cell Lymphoma In Vivo. J. Immunol. 2016, 196, 4805–4813. [Google Scholar] [CrossRef] [Green Version]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, L.; Zong, J.; Ye, Y.; Chen, G.; Chen, Y.; Liao, X.; Guo, Q.; Qiu, S.; Lin, S.; et al. Decreased expression of the NKG2D ligand ULBP4 may be an indicator of poor prognosis in patients with nasopharyngeal carcinoma. Oncotarget 2017, 8, 42007–42019. [Google Scholar] [CrossRef]
- McGilvray, R.W.; Eagle, R.A.; Watson, N.F.S.; Al-Attar, A.; Ball, G.; Jafferji, I.; Trowsdale, J.; Durrant, L.G. NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin. Cancer Res. 2009, 15, 6993–7002. [Google Scholar] [CrossRef]
- Park, Y.P.; Choi, S.-C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the γc cytokines and TGF-β1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Yim, D.; Chow, I.-T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lundgren, A.D.; Singh, P.; Goodlett, D.R.; Plymate, S.R.; Wu, J.D. An six-amino acid motif in the alpha3 domain of MICA is the cancer therapeutic target to inhibit shedding. Biochem. Biophys. Res. Commun. 2009, 387, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, J.; Liu, D.; Li, G.; Staveley-O’Carroll, K.F.; Li, Z.; Wu, J.D. Nonblocking Monoclonal Antibody Targeting Soluble MIC Revamps Endogenous Innate and Adaptive Antitumor Responses and Eliminates Primary and Metastatic Tumors. Clin. Cancer Res. 2015, 21, 4819–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weil, S.; Memmer, S.; Lechner, A.; Huppert, V.; Giannattasio, A.; Becker, T.; Müller-Runte, A.; Lampe, K.; Beutner, D.; Quaas, A.; et al. Natural Killer Group 2D Ligand Depletion Reconstitutes Natural Killer Cell Immunosurveillance of Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2017, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, D.; Li, G.; Staveley-O’Carroll, K.F.; Graff, J.N.; Li, Z.; Wu, J.D. Antibody-mediated neutralization of soluble MIC significantly enhances CTLA4 blockade therapy. Sci. Adv. 2017, 3, e1602133. [Google Scholar] [CrossRef] [Green Version]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef]
- Wiemann, K.; Mittrücker, H.-W.; Feger, U.; Welte, S.A.; Yokoyama, W.M.; Spies, T.; Rammensee, H.-G.; Steinle, A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J. Immunol. 2005, 175, 720–729. [Google Scholar] [CrossRef]
- Thompson, T.W.; Kim, A.B.; Li, P.J.; Wang, J.; Jackson, B.T.; Huang, K.T.H.; Zhang, L.; Raulet, D.H. Endothelial cells express NKG2D ligands and desensitize antitumor NK responses. Elife 2017, 6, e30881. [Google Scholar] [CrossRef]
- Deng, W.; Gowen, B.G.; Zhang, L.; Wang, L.; Lau, S.; Iannello, A.; Xu, J.; Rovis, T.L.; Xiong, N.; Raulet, D.H. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015, 348, 136–139. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentman, C.L.; Meehan, K.R. NKG2D CARs as cell therapy for cancer. Cancer J. 2014, 20, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Zhang, T.; Megli, C.J.; Wu, J.; Meehan, K.R.; Sentman, C.L. Chimeric NKG2D receptor-expressing T cells as an immunotherapy for multiple myeloma. Exp. Hematol. 2008, 36, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.; Zhang, T.; DeMars, L.R.; Conejo-Garcia, J.; Roby, K.F.; Sentman, C.L. Chimeric NKG2D receptor-bearing T cells as immunotherapy for ovarian cancer. Cancer Res. 2007, 67, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Metais, J.-Y.; Escudero, A.; Vela, M.; Valentín, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patiño-García, A.; et al. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef]
- Han, Y.; Xie, W.; Song, D.-G.; Powell, D.J. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef]
- Chang, Y.-H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Lonez, C.; Verma, B.; Hendlisz, A.; Aftimos, P.; Awada, A.; Van Den Neste, E.; Catala, G.; Machiels, J.-P.H.; Piette, F.; Brayer, J.B.; et al. Study protocol for THINK: A multinational open-label phase I study to assess the safety and clinical activity of multiple administrations of NKR-2 in patients with different metastatic tumour types. BMJ Open 2017, 7, e017075. [Google Scholar] [CrossRef]
- Davis, Z.B.; Vallera, D.A.; Miller, J.S.; Felices, M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin. Immunol. 2017, 31, 64–75. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target Oncol. 2016, 11, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol. Biol. 2016, 1441, 333–346. [Google Scholar] [PubMed] [Green Version]
- Schmohl, J.U.; Felices, M.; Oh, F.; Lenvik, A.J.; Lebeau, A.M.; Panyam, J.; Miller, J.S.; Vallera, D.A. Engineering of Anti-CD133 Trispecific Molecule Capable of Inducing NK Expansion and Driving Antibody-Dependent Cell-Mediated Cytotoxicity. Cancer Res. Treat. 2017, 49, 1140–1152. [Google Scholar] [CrossRef] [Green Version]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunological Mechanisms Underneath the Efficacy of Cancer Therapy. Cancer Immunol. Res. 2016, 4, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.-C.; Ho, Y.-J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; DeFor, T.E.; et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018, 131, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Rosario, M.; Romee, R.; Berrien-Elliott, M.M.; Schneider, S.E.; Leong, J.W.; Sullivan, R.P.; Jewell, B.A.; Becker-Hapak, M.; Schappe, T.; et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Invest. 2017, 127, 4042–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.-G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Barrow, A.D.; Colonna, M. Tailoring Natural Killer cell immunotherapy to the tumour microenvironment. Semin. Immunol. 2017, 31, 30–36. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrow, A.D.; Colonna, M. Exploiting NK Cell Surveillance Pathways for Cancer Therapy. Cancers 2019, 11, 55. https://doi.org/10.3390/cancers11010055
Barrow AD, Colonna M. Exploiting NK Cell Surveillance Pathways for Cancer Therapy. Cancers. 2019; 11(1):55. https://doi.org/10.3390/cancers11010055
Chicago/Turabian StyleBarrow, Alexander David, and Marco Colonna. 2019. "Exploiting NK Cell Surveillance Pathways for Cancer Therapy" Cancers 11, no. 1: 55. https://doi.org/10.3390/cancers11010055
APA StyleBarrow, A. D., & Colonna, M. (2019). Exploiting NK Cell Surveillance Pathways for Cancer Therapy. Cancers, 11(1), 55. https://doi.org/10.3390/cancers11010055