Parathyroid Cancer: A Review

Abstract
1. Introduction
2. Epidemiology
3. Etiology and Pathogenesis
4. Clinical Presentation
- A higher frequency of symptomatic hypercalcemia. These patients can present with a myriad of symptoms, including nausea, vomiting, abdominal pain, constipation, fatigue, myopathy, disorientation and neurocognitive deficits;
- Very high serum PTH concentrations (5–10 × the upper limit of normal, as well as absolute PTH levels >500 mg/dL;
- Serum calcium levels >14 mg/dL;
- Presence of a parathyroid crisis;
- Presence of a palpable neck mass.
4.1. Atypical Presentations
4.2. Hypercalcemic Crisis
- Normal saline is the intravenous fluid of choice for resuscitation and volume expansion. The initial rate of administration is between 200 and 300 cc/hr, which is titrated to ensure a urine output of 100–150 cc/hr [25].
- While loop diuretics aid with calcium excretion, they are not usually recommended in the absence of cardiac or renal failure, since there are possible complications and due to the presence of alternative medications that inhibit bone resorption (usually are the main proponent of hypercalcemia);
- Another effective medication is salmon calcitonin (4 international units/kg) with repeat serum measurements a few hours later. The goal is to evaluate for an appropriate decrease in calcium level, at which time repeat doses can be given for 6–12 h (4–8 IU/hour). It is important to note that sometimes patients develop tachyphylaxis to this medication after repeated doses, therefore the medication is not usually continued beyond 24–48 h [26];
- Another class of medications that are useful include bisphosphonates. These medications are non-hydrolysable compounds that adsorb to bone surfaces and inhibit calcium release by interfering with bone resorption [27];
- Zolendronate (4 mg IV over 15 min) or pamidronate (60–90 mg/2 h) can also be administered, with repeat doses as needed every 3–4 weeks. Zolendronate has been found to be more effective in cases of malignancy induced hypercalcemia than pamidronate;
- Certain patients are unable to receive bisphosphonates due to renal impairment. In these cases, a medication called denosumab may be administered instead, in addition to calcitonin. The initial dose is 60 mg subcutaneously, repeated for clinical response. [28]. Recent studies even support a higher dose regimen of 120 mg of denosumab, given every 4 weeks, which has shown to be very effective in controlling hypercalcemia from bone metastases in advanced cancer [29]. Another study included doses on day 8 and 15 during the first month, to expedite the drop in calcium and achieve a steady state of denosumab at a faster rate [30].
5. Diagnosis
- Sheets or lobules of tumor cells with interspersed fibrous bands;
- Mitotic figures;
- Necrosis;
- Capsular invasion;
- Vascular invasion.
5.1. Images Courtesy of Sylvia L. Asa, Dept. of Pathology, Case Western Reserve University, OH
5.2. Atypical Adenomas
6. Staging
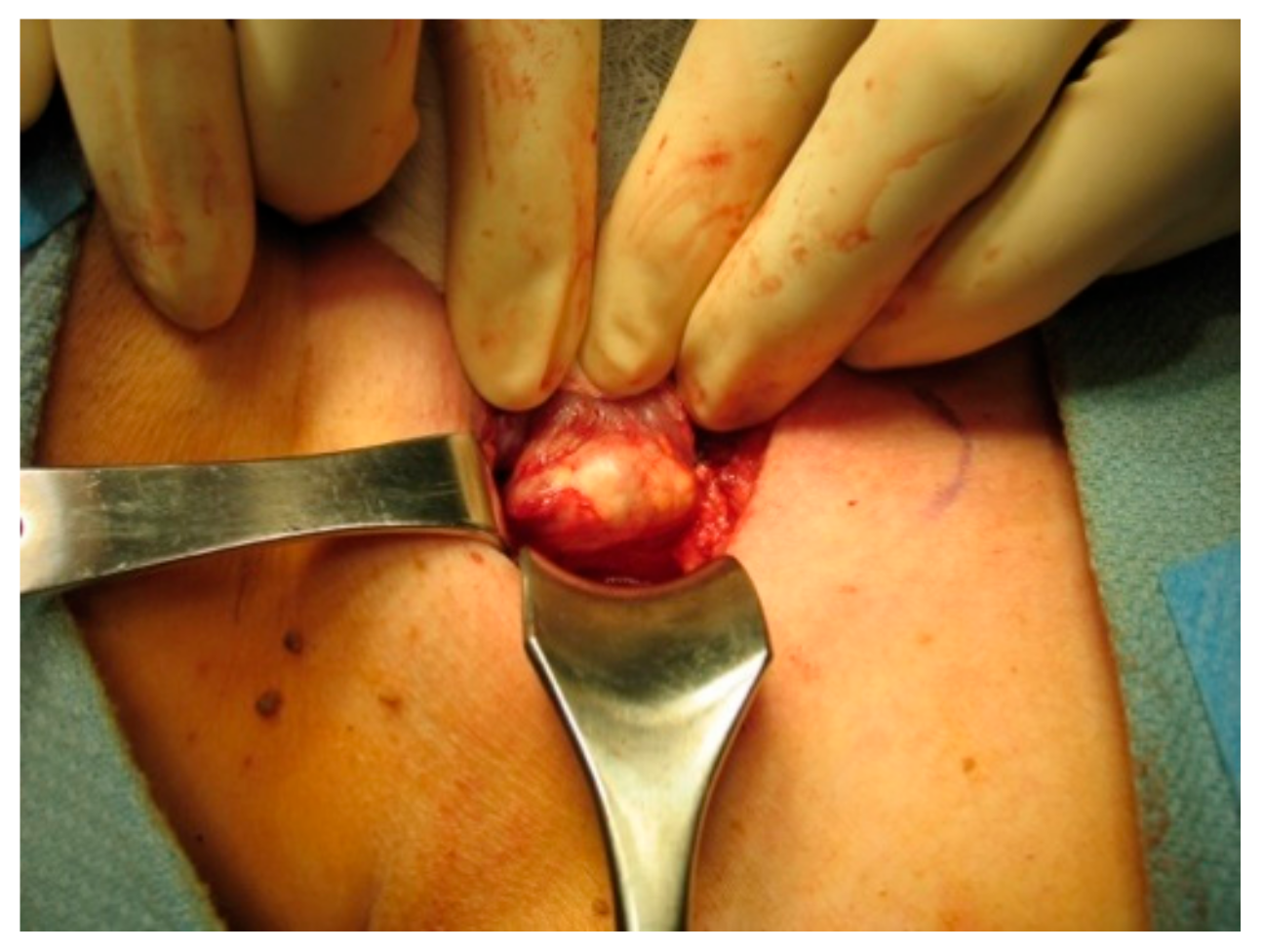
7. Surgical Treatment of Parathyroid Cancer
7.1. Preoperative Imaging
7.2. Operative Intervention
Principles of en Bloc Surgery
- A complete and thorough exploration of all four glands will help identify the presence of concurrent adenomas and carcinomas. While rare, multiglandular carcinomas have been described in the literature [54];
- A bloodless field and meticulous attention to detail ensures no inadvertent injury to neighboring structures;
- Careful inspection for any tumor encroachment into the surrounding strap muscles or other structures, which may have to be resected with the mass. The most common structures affected by local tumor invasion are the ipsilateral thyroid lobe, ipsilateral strap muscles, ipsilateral recurrent laryngeal nerve, esophagus, and trachea [56];
- Nodal involvement necessitates a regional lymph node dissection of that compartment. It is important to note that prophylactic lateral neck dissection has not been shown to improve survival, is associated with an increased morbidity, and is therefore not recommended [57];
- In most cases, the recurrent laryngeal nerve can and should be preserved. However, if there is evidence of RLN involvement, it may be sacrificed and resected with the tumor [58].
7.3. Post-Operative Management
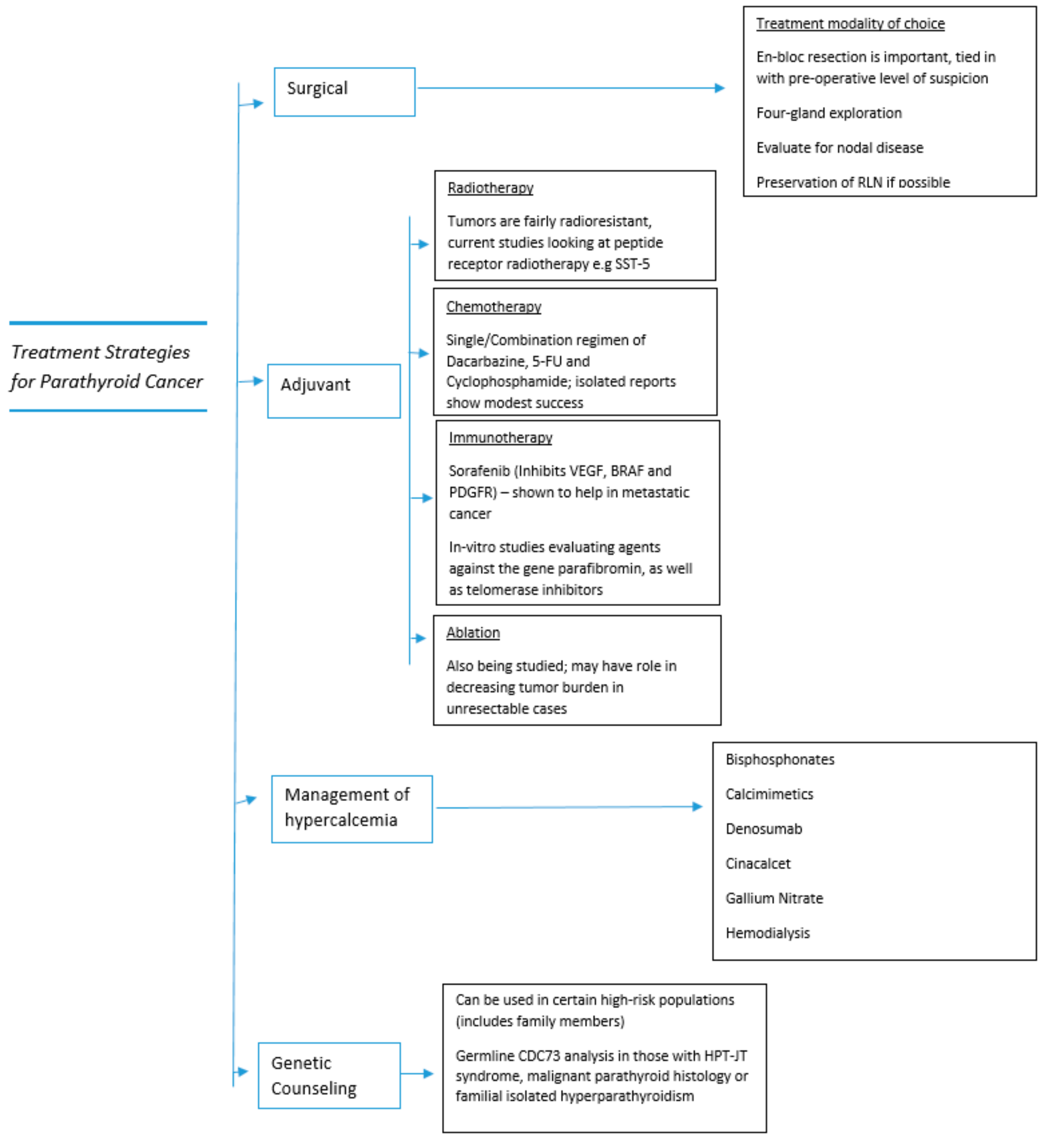
8. Adjuvant Therapy for Parathyroid Cancer
8.1. Radiotherapy
8.2. Chemotherapy
8.3. Immunotherapy
8.4. Agents for Symptom Palliation/Management of Hypercalcemia
8.5. Newer Treatment Modalities
9. Management of Recurrent Disease
10. Survival and Outcomes
11. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Fraser, W.D. Hyperparathyroidism. Lancet 2009, 374, 145–158. [Google Scholar] [CrossRef]
- Marcocci, C.; Cetani, F. Primary Hyperparathyroidism. N. Engl. J. Med. 2011, 365, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Sainton, P.M.J. Malegne d’un adenoma parathyroidiene eosinophile [Malignant eosinophilic parathyroid] Au cours d’une de Recklinghausen. Ann. Ann. Pathol. 1933, 10, 813. [Google Scholar]
- Lee, P.K.; Jarosek, S.L.; Virnig, B.A.; Evasovich, M.; Tuttle, T.M. Trends in the incidence and treatment of parathyroid cancer in the United States. Cancer 2007, 109, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, C.; Cetani, F.; Rubin, M.R.; Silverberg, S.J.; Pinchera, A.; Bilezikian, J.P. Parathyroid carcinoma. J. Bone Miner. Res. 2008, 23, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- McMullen, T.; Bodie, G.; Gill, A.; Ihre-Lundgren, C.; Shun, A.; Bergin, M.; Stevens, G.; Delbridge, L. Hyperparathyroidism After Irradiation for Childhood Malignancy. Int. J. Radiat. Oncol. 2009, 73, 1164–1168. [Google Scholar] [CrossRef]
- Rasmuson, T.; Damber, L.; Johansson, L.; Johansson, R.; Larsson, L.-G. Increased incidence of parathyroid adenomas following X-ray treatment of benign diseases in the cervical spine in adult patients. Clin. Endocrinol. (Oxf.) 2002, 57, 731–734. [Google Scholar] [CrossRef]
- Pimenta, F.J.; Gontijo Silveira, L.F.; Tavares, G.C.; Silva, A.C.; Perdigão, P.F.; Castro, W.H.; Gomez, M.V.; Teh, B.T.; Marco, L.; Gomez, R.S. HRPT2 gene alterations in ossifying fibroma of the jaws. Oral Oncol. 2006, 42, 735–739. [Google Scholar] [CrossRef]
- Chen, J.D.; Morrison, C.; Zhang, C.; Kahnoski, K.; Carpten, J.D.; Teh, B.T. Hyperparathyroidism-jaw tumour syndrome. J. Intern. Med. 2003, 253, 634–642. [Google Scholar] [CrossRef]
- Bradley, K.J.; Hobbs, M.R.; Buley, I.D.; Carpten, J.D.; Cavaco, B.M.; Fares, J.E.; Laidler, P.; Manek, S.; Robbins, C.S.; Salti, I.S.; et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J. Intern. Med. 2005, 257, 18–26. [Google Scholar] [CrossRef]
- Givi, B.; Shah, J.P. Parathyroid carcinoma. Clin. Oncol (R Coll Radiol) 2010, 22, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Carpten, J.D.; Robbins, C.M.; Villablanca, A.; Forsberg, L.; Presciuttini, S.; Bailey-Wilson, J.; Simonds, W.F.; Gillanders, E.M.; Kennedy, A.M.; Chen, J.D.; et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism–jaw tumor syndrome. Nat. Genet. 2002, 32, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Howell, V.M.; Haven, C.J.; Kahnoski, K.; Khoo, S.K.; Petillo, D.; Chen, J.; Fleuren, G.J.; Robinson, B.G.; Delbridge, L.W.; Philips, J.; et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J. Med. Genet. 2003, 40, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.J. New Concepts About Familial Isolated Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2019, 104, 4058. [Google Scholar] [CrossRef] [PubMed]
- Cetani, F.; Pardi, E.; Ambrogini, E.; Lemmi, M.; Borsari, S.; Cianferotti, L.; Vignali, E.; Viacava, P.; Berti, P.; Mariotti, S.; et al. Genetic analyses in familial isolated hyperparathyroidism: Implication for clinical assessment and surgical management. Clin. Endocrinol. (Oxf.) 2006, 64, 146–152. [Google Scholar] [CrossRef]
- Arnold, A.; Shattuck, T.M.; Mallya, S.M.; Krebs, L.J.; Costa, J.; Gallagher, J.; Wild, Y.; Saucier, K. Molecular pathogenesis of primary hyperparathyroidism. J. Bone Miner. Res. 2002, 17 (Suppl. 2), N30–N36. [Google Scholar] [PubMed]
- Cardoso, L.; Stevenson, M.; Thakker, R.V. Molecular genetics of syndromic and non-syndromic forms of parathyroid carcinoma. Hum. Mutat. 2017, 38, 1621–1648. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, X.; Cui, M.; Wang, M.; Su, Z.; Liao, Q.; Zhao, Y. Circular RNA profile of parathyroid neoplasms: Analysis of co-expression networks of circular RNAs and mRNAs. RNA Biol. 2019, 16, 1228–1236. [Google Scholar] [CrossRef]
- Clarke, C.N.; Katsonis, P.; Hsu, T.-K.; Koire, A.M.; Silva-Figueroa, A.; Christakis, I.; Williams, M.; Kutahyalioglu, M.; Kwatampora, L.; Xi, Y.; et al. Comprehensive Genomic Characterization of Parathyroid Cancer Identifies Novel Candidate Driver Mutations and Core Pathways. J. Endocr. Soc. 2019, 3, 544–559. [Google Scholar] [CrossRef]
- Kang, H.; Pettinga, D.; Schubert, A.D.; Ladenson, P.W.; Ball, D.W.; Chung, J.H.; Shrock, A.; Madison, R.; Frampton, G.; Stephens, P.; et al. Genomic Profiling of Parathyroid Carcinoma Reveals Genomic Alterations Suggesting Benefit from Therapy. Oncologist 2019, 24, 791–797. [Google Scholar] [CrossRef]
- Messerer, C.L.; Bugis, S.P.; Baliski, C.; Wiseman, S.M. Normocalcemic parathyroid carcinoma: An unusual clinical presentation. World J. Surg. Oncol. 2006, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, D.; Chen, W.; Zhang, S.; Wang, Z.; Li, K.; Gao, Y.; Zou, S.; Yang, A. Non-functional parathyroid carcinoma: A case report and review of the literature. Cancer Biol. Ther. 2015, 16, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ranvier, G.; Jensen, K.; Khanafshar, E.; Quivey, J.; Glastonbury, C.; Kebebew, E.; Duh, Q.-Y.; Clark, O. Nonfunctioning Parathyroid Carcinoma: Case Report and Review of Literature. Endocr. Pract. 2007, 13, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, S.E.; Perrier, N.D. Parathyroid carcinoma. Curr. Opin. Oncol. 2006, 18, 16–22. [Google Scholar] [CrossRef] [PubMed]
- LeGrand, S.B. Modern Management of Malignant Hypercalcemia. Am. J. Hosp. Palliat. Med. 2011, 28, 515–517. [Google Scholar] [CrossRef]
- Mirrakhimov, A.E. Hypercalcemia of Malignancy: An Update on Pathogenesis and Management. N. Am. J. Med. Sci. 2015, 7, 483–493. [Google Scholar] [CrossRef]
- Sternlicht, H.; Glezerman, I.G. Hypercalcemia of malignancy and new treatment options. Ther. Clin. Risk Manag. 2015, 11, 1779–1788. [Google Scholar]
- Dietzek, A.; Connelly, K.; Cotugno, M.; Bartel, S.; McDonnell, A.M. Denosumab in hypercalcemia of malignancy: A case series. J. Oncol. Pharm. Pract. 2015, 21, 143–147. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; De Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Hu, M.I.; Glezerman, I.G.; Leboulleux, S.; Insogna, K.; Gucalp, R.; Misiorowski, W.; Yu, B.; Zorsky, P.; Tosi, D.; Bessudo, A.; et al. Denosumab for treatment of hypercalcemia of malignancy. J. Clin. Endocrinol. Metab. 2014, 99, 3144–3152. [Google Scholar] [CrossRef]
- MMBasso, S.; Lumachi, F.; Nascimben, F.; Luisetto, G.; Camozzi, V. Treatment of Acute Hypercalcemia. Med. Chem. (Los Angeles) 2012, 8, 564–568. [Google Scholar]
- Sharretts, J.M.; Kebebew, E.; Simonds, W.F. Parathyroid Cancer. Semin. Oncol. 2010, 37, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Al-Kurd, A.; Mekel, M.; Mazeh, H. Parathyroid carcinoma. Surg. Oncol. 2014, 23, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Bancos, I.; Grant, C.; Nadeem, S.; Stan, M.; Reading, C.; Sebo, T.; Algeciras-Simnich, A.; Singh, R.; Dean, D. Risks and Benefits of Parathyroid Fine-Needle Aspiration with Parathyroid Hormone Washout. Endocr. Pract. 2012, 18, 441–449. [Google Scholar] [CrossRef]
- Kim, J.; Horowitz, G.; Hong, M.; Orsini, M.; Asa, S.L.; Higgins, K. The dangers of parathyroid biopsy. J. Otolaryngol. Head Neck Surg. 2017, 46, 4. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Wang, T.S.; Ruan, D.T.; Lee, J.A.; Asa, S.L.; Duh, Q.-Y.; Doherty, G.; Herrera, M.; Pasieka, J.; Perrier, N.; et al. The American Association of Endocrine Surgeons Guidelines for Definitive Management of Primary Hyperparathyroidism. JAMA Surg. 2016, 151, 959–968. [Google Scholar] [CrossRef]
- Schantz, A.; Castleman, B. Parathyroid carcinoma. A study of 70 cases. Cancer 1973, 31, 600–605. [Google Scholar] [CrossRef]
- Quinn, C.E.; Healy, J.; Lebastchi, A.H.; Brown, T.C.; Stein, J.E.; Prasad, M.L.; Callender, G.; Carling, T.; Udelsman, R. Modern Experience with Aggressive Parathyroid Tumors in a High-Volume New England Referral Center. J. Am. Coll. Surg. 2015, 220, 1054–1062. [Google Scholar] [CrossRef]
- Truran, P.P.; Johnson, S.J.; Bliss, R.D.; Lennard, T.W.J.; Aspinall, S.R. Parafibromin, Galectin-3, PGP9.5, Ki67, and Cyclin D1: Using an Immunohistochemical Panel to Aid in the Diagnosis of Parathyroid Cancer. World J. Surg. 2014, 38, 2845–2854. [Google Scholar] [CrossRef]
- Erovic, B.M.; Harris, L.; Jamali, M.; Goldstein, D.P.; Irish, J.C.; Asa, S.L.; Ozgur, M. Biomarkers of Parathyroid Carcinoma. Endocr. Pathol. 2012, 23, 221–231. [Google Scholar] [CrossRef]
- Fernandez-Ranvier, G.G.; Khanafshar, E.; Jensen, K.; Zarnegar, R.; Lee, J.; Kebebew, E.; Duh, Q.-Y.; Clark, O. Parathyroid carcinoma, atypical parathyroid adenoma, or parathyromatosis? Cancer 2007, 110, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Long, K.L.; Sippel, R.S. Current and future treatments for parathyroid carcinoma. Int. J. Endocr. Oncol. 2018, 5, IJE06. [Google Scholar] [CrossRef]
- Landry, C.S.; Wang, T.S.; Asare, A.E.; Grogan, R.; Hunt, J.; Ridge, J.A.; Rohren, E.; Shah, J.P.; Subramaniam, R.M.; Brierley, J.D.; et al. Parathyroid in AJCC; Springer: New York, NY, USA, 2017; p. 903. [Google Scholar]
- Sidhu, P.S.; Talat, N.; Patel, P.; Mulholland, N.J.; Schulte, K.-M. Ultrasound features of malignancy in the preoperative diagnosis of parathyroid cancer: A retrospective analysis of parathyroid tumours larger than 15 mm. Eur. Radiol. 2011, 21, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Abboud, B.; Sleilaty, G.; Rabaa, L.; Daher, R.; Zeid, H.A.; Jabbour, H.; Hachem, K.; Smayra, T. Ultrasonography: Highly Accuracy Technique for Preoperative Localization of Parathyroid Adenoma. Laryngoscope 2008, 118, 1574–1578. [Google Scholar] [CrossRef] [PubMed]
- Lavely, W.C.; Goetze, S.; Friedman, K.P.; Leal, J.P.; Zhang, Z.; Garret-Mayer, E.; Dackiw, A.; Tufano, R.; Zeiger, M.; Zeissman, H. Comparison of SPECT/CT, SPECT, and planar imaging with single- and dual-phase (99m)Tc-sestamibi parathyroid scintigraphy. J. Nucl. Med. 2007, 48, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Hetrakul, N.; Civelek, A.C.; Stagg, C.A.; Udelsman, R. In vitro accumulation of technetium-99m-sestamibi in human parathyroid mitochondria. Surgery 2001, 130, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; Yip, L.; Tublin, M.E. Cystic Parathyroid Adenoma: Sonographic Features and Correlation With 99m Tc-Sestamibi SPECT Findings. Am. J. Roentgenol. 2010, 195, 1385–1390. [Google Scholar] [CrossRef]
- Pappu, S.; Donovan, P.; Cheng, D.; Udelsman, R. Sestamibi scans are not all created equally. Arch. Surg. 2005, 140, 383–386. [Google Scholar] [CrossRef]
- Kunstman, J.W.; Kirsch, J.D.; Mahajan, A.; Udelsman, R. Parathyroid Localization and Implications for Clinical Management. J. Clin. Endocrinol. Metab. 2013, 98, 902–912. [Google Scholar] [CrossRef]
- Weber, A.L.; Randolph, G.; Aksoy, F.G. The thyroid and parathyroid glands. CT and MR imaging and correlation with pathology and clinical findings. Radiol. Clin. North. Am. 2000, 38, 1105–1129. [Google Scholar] [CrossRef]
- Poeppel, T.D.; Krause, B.J.; Heusner, T.A.; Boy, C.; Bockisch, A.; Antoch, G. PET/CT for the staging and follow-up of patients with malignancies. Eur. J. Radiol. 2009, 70, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, L.; Sorgato, N.; Torresan, F.; Boschin, I.M.; Pennelli, G.; Saladini, G.; Piotto, A.; Rubello, D.; Pelizzo, M.R. FDG-PET/CT and parathyroid carcinoma: Review of literature and illustrative case series. World J. Clin. Oncol. 2011, 2, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Sahasranam, P.; Tran, M.T.; Mohamed, H.; Friedman, T.C. Multiglandular Parathyroid Carcinoma: A Case Report and Brief Review. South. Med. J. 2007, 100, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E. Parathyroid carcinoma. Curr. Treat. Options Oncol. 2001, 2, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hoelting, T.; Weber, T.; Werner, J.; Herfarth, C. Surgical treatment of parathyroid carcinoma (Review). Oncol. Rep. 2001, 8, 931–934. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sandelin, K.; Auer, G.; Bondeson, L.; Grimelius, L.; Farnebo, L.O. Prognostic factors in parathyroid cancer: A review of 95 cases. World J. Surg. 1992, 16, 724–731. [Google Scholar] [CrossRef]
- Kebebew, E.; Arici, C.; Duh, Q.Y.; Clark, O.H. Localization and reoperation results for persistent and recurrent parathyroid carcinoma. Arch. Surg. 2001, 136, 878–885. [Google Scholar] [CrossRef]
- Ippolito, G.; Palazzo, F.F.; Sebag, F.; De Micco, C.; Henry, J.F. Intraoperative diagnosis and treatment of parathyroid cancer and atypical parathyroid adenoma. Br. J. Surg. 2007, 94, 566–570. [Google Scholar] [CrossRef]
- Dobrinja, C.; Santandrea, G.; Giacca, M.; Stenner, E.; Ruscio, M.; de Manzini, N. Effectiveness of Intraoperative Parathyroid Monitoring (ioPTH) in predicting a multiglandular or malignant parathyroid disease. Int. J. Surg. 2017, 41, S26–S33. [Google Scholar] [CrossRef]
- Schulte, K.-M.; Gill, A.J.; Barczynski, M.; Karakas, E.; Miyauchi, A.; Knoefel, W.T.; Lombardi, C.P.; Talat, N.; Diaz-Cano, S.; Grant, C.S. Classification of Parathyroid Cancer. Ann. Surg. Oncol. 2012, 19, 2620–2628. [Google Scholar] [CrossRef]
- Talat, N.; Schulte, K.-M. Clinical Presentation, Staging and Long-Term Evolution of Parathyroid Cancer. Ann. Surg. Oncol. 2010, 17, 2156–2174. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-T.; Sippel, R.S.; Chen, H.; Schneider, D.F. Is central lymph node dissection necessary for parathyroid carcinoma? Surgery 2014, 156, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Agnes, N.; Hamdy, T.; Witteveen, J.E.; Van Thiel, S.; Romijn, J.A.; Hamdy, N.A.T. Therapy of Endocrine Disease: Hungry bone syndrome: Still a challenge in the post-operative management of primary hyperparathyroidism: A systematic review of the literature Hungry bone syndrome: Still a challenge in the post-operative management of primary hyperparathyroidism: A systematic review of the literature. Artic Eur. J. Endocrinol. 2012, 168, 45–53. [Google Scholar]
- Busaidy, N.L.; Jimenez, C.; Habra, M.A.; Schultz, P.N.; El-Naggar, A.K.; Clayman, G.L.; Asper, J.; Diaz, E.M.; Evans, D.; Gagel, R.; et al. Parathyroid carcinoma: A 22-year experience. Head Neck 2004, 26, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Munson, N.D.; Foote, R.L.; Northcutt, R.C.; Tiegs, R.D.; Fitzpatrick, L.A.; Grant, C.S.; vanHeerden, J.; Thompson, G.; Lloyd, R.V. Parathyroid carcinoma: Is there a role for adjuvant radiation therapy? Cancer 2003, 98, 2378–2384. [Google Scholar] [CrossRef]
- Erovic, B.M.; Goldstein, D.P.; Kim, D.; Mete, O.; Brierley, J.; Tsang, R.; Freeman, J.L.; Asa, S.L.; Rotstein, L.; Irish, J.C. Parathyroid cancer: Outcome analysis of 16 patients treated at the princess margaret hospital. Head Neck 2013, 35, 35–39. [Google Scholar] [CrossRef]
- Storvall, S.; Leijon, H.; Rhyanen, E.; Louhimo, J.; Haglund, C.; Schalin-Jantti, A.J. Somatostatin receptor expression in parathyroid neoplasms. Endocr. Connect. 2019, 8, 1213–1223. [Google Scholar] [CrossRef]
- Shane, E. Parathyroid Carcinoma. J. Clin. Endocrinol. Metab. 2001, 86, 485–493. [Google Scholar] [CrossRef]
- Owen, R.P.; Silver, C.E.; Pellitteri, P.K.; Shaha, A.R.; Devaney, K.O.; Werner, J.A.; Rinaldo, A.; Ferlito, A. Parathyroid carcinoma: A review. Head Neck 2010, 33, 429–436. [Google Scholar] [CrossRef]
- Betea, D.; Bradwell, A.R.; Harvey, T.C.; Mead, G.P.; Schmidt-Gayk, H.; Ghaye, B.; Daly, A.F.; Beckers, A. Hormonal and Biochemical Normalization and Tumor Shrinkage Induced by Anti-Parathyroid Hormone Immunotherapy in a Patient with Metastatic Parathyroid Carcinoma. J. Clin. Endocrinol. Metab. 2004, 89, 3413–3420. [Google Scholar] [CrossRef]
- Alharbi, N.; Asa, S.L.; Szybowska, M.; Kim, R.H.; Ezzat, S. Intrathyroidal Parathyroid Carcinoma: An Atypical Thyroid Lesion. Front. Endocrinol. (Lausanne) 2018, 9, 641. [Google Scholar] [CrossRef]
- Rozhinskaya, L.; Pigarova, E.; Sabanova, E.; Mamedova, E.; Voronkova, I.; Krupinova, J.; Dzeranova, L.; Tiulpakov, A.; Gorbunova, V.; Orel, N.; et al. Diagnosis and treatment challenges of parathyroid carcinoma in a 27-year-old woman with multiple lung metastases. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Fountas, A.; Andrikoula, M.; Giotaki, Z.; Limniati, C.; Tsakiridou, E.; Tigas, S.; Tsatsoulis, A. The Emerging Role of Denosumab in the Long-Term Management of Parathyroid Carcinoma-Related Refractory Hypercalcemia. Endocr. Pract. 2015, 21, 468–473. [Google Scholar] [CrossRef]
- Karuppiah, D.; Thanabalasingham, G.; Shine, B.; Wang, L.M.; Sadler, G.P.; Karavitaki, N.; Grossman, A. Refractory hypercalcaemia secondary to parathyroid carcinoma: Response to high-dose denosumab. Eur. J. Endocrinol. 2014, 171, K1–K5. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Brown, J.E.; Van Poznak, C.; Ibrahim, T.; Stemmer, S.M.; Stopeck, A.T.; Diel, I.J.; Takahashi, S.; Shore, N.; Henry, D.H.; et al. Incidence, risk factors, and outcomes of osteonecrosis of the jaw: Integrated analysis from three blinded active-controlled phase III trials in cancer patients with bone metastases. Ann. Oncol. 2012, 23, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, S.J.; Rubin, M.R.; Faiman, C.; Peacock, M.; Shoback, D.M.; Smallridge, R.C.; Schwanauer, L.E.; Olson, K.A.; Klassen, P.; Bilezikian, J.P. Cinacalcet Hydrochloride Reduces the Serum Calcium Concentration in Inoperable Parathyroid Carcinoma. J. Clin. Endocrinol. Metab. 2007, 92, 3803–3808. [Google Scholar] [CrossRef] [PubMed]
- Cvitkovic, F.; Armand, J.-P.; Tubiana-Hulin, M.; Rossi, J.-F.; Warrell, R.P. Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J. 2006, 12, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Montenegro FL de, M.; Chammas, M.C.; Juliano, A.G.; Cernea, C.R.; Cordeiro, A.C. Ethanol injection under ultrasound guidance to palliate unresectable parathyroid carcinoma. Arq. Bras. Endocrinol. Metabol. 2008, 52, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Seehofer, D.; Steinmüller, T.; Rayes, N.; Podrabsky, P.; Riethmüller, J.; Klupp, J.; Ulrich, F.; Schindler, R.; Frei, U.; Neuhaus, P. Parathyroid Hormone Venous Sampling Before Reoperative Surgery in Renal Hyperparathyroidism. Arch. Surg. 2004, 139, 1331. [Google Scholar] [CrossRef]
- Mezhir, J.J.; Melis, M.; Headley, R.C.; Pai, R.K.; Posner, M.C.; Kaplan, E.L. Successful palliation of hypercalcemia secondary to metastatic parathyroid cancer: An unusual indication for hepatic resection. J. Hepatobiliary Pancreat Surg. 2007, 14, 410–413. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; McHenry, C.R. Parathyroid carcinoma. J. Surg. Oncol. 2005, 89, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.-M.; Talat, N.; Miell, J.; Moniz, C.; Sinha, P.; Diaz-Cano, S. Lymph Node Involvement and Surgical Approach in Parathyroid Cancer. World J. Surg. 2010, 34, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Asare, E.A.; Sturgeon, C.; Winchester, D.J.; Liu, L.; Palis, B.; Perrier, N.D.; Evans, D.; Winchester, D.; Wang, T. Parathyroid Carcinoma: An Update on Treatment Outcomes and Prognostic Factors from the National Cancer Data Base (NCDB). Ann. Surg. Oncol. 2015, 22, 3990–3995. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Simonds, W.F. Endocrine neoplasms in familial syndromes of hyperparathyroidism. Endocr. Relat. Cancer 2016, 23, R229–R247. [Google Scholar] [CrossRef][Green Version]
- Van der Tuin, K.; Tops, C.M.J.; Adank, M.A.; Cobben, J.-M.; Hamdy, N.A.T.; Jongmans, M.C.; Menko, F.H.; van Nesselroojj, B.P.M.; Netea-Maier, R.; Oosterwijk, J.C.; et al. CDC73-Related Disorders: Clinical Manifestations and Case Detection in Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 4534–4540. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normocalcemic (Serum Ca < 10.5 mg/dL) | Hypercalcemic | Hypercalcemic Crisis (Serum Ca > 14 mg/dL) |
|---|---|---|
| Can be asymptomatic, may have palpable mass | Usually symptomatic, gastrointestinal disturbances, renal stones or fatigue, neurocognitive issues | Can present in extremis, with altered sensorium or stupor |
| Present at more advanced stages | Should have a high index of suspicion based on calcium and PTH levels | Aggressive management with hydration, bisphosphonates, calcitonin or denosumab |
| Higher tendency to metastasize | Expedite surgery |
| Patient Factors | Tumor Related Factors | Lab/Histological Factors |
|---|---|---|
| Age at diagnosis | Size of primary tumor | Highest preoperative calcium |
| Gender | Presence of invasion into surrounding tissue | Highest preoperative PTH |
| Race | Distant metastatic disease | + lymphovascular invasion |
| Genetic mutations | Number of lymph nodes removed | Histological grade (high or low) and Ki67 index |
| Number of positive lymph nodes | Mitotic rate | |
| Weight of primary tumor | Solid vs. trabecular growth pattern | |
| Time to recurrence | Tumor necrosis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, N.N.; Wilhelm, S.M. Parathyroid Cancer: A Review. Cancers 2019, 11, 1676. https://doi.org/10.3390/cancers11111676
Machado NN, Wilhelm SM. Parathyroid Cancer: A Review. Cancers. 2019; 11(11):1676. https://doi.org/10.3390/cancers11111676
Chicago/Turabian StyleMachado, Nikita N, and Scott M Wilhelm. 2019. "Parathyroid Cancer: A Review" Cancers 11, no. 11: 1676. https://doi.org/10.3390/cancers11111676
APA StyleMachado, N. N., & Wilhelm, S. M. (2019). Parathyroid Cancer: A Review. Cancers, 11(11), 1676. https://doi.org/10.3390/cancers11111676