Models for Understanding Resistance to Chemotherapy in Liver Cancer

 ,
,  ,
,  and
and 
Abstract
1. Introduction
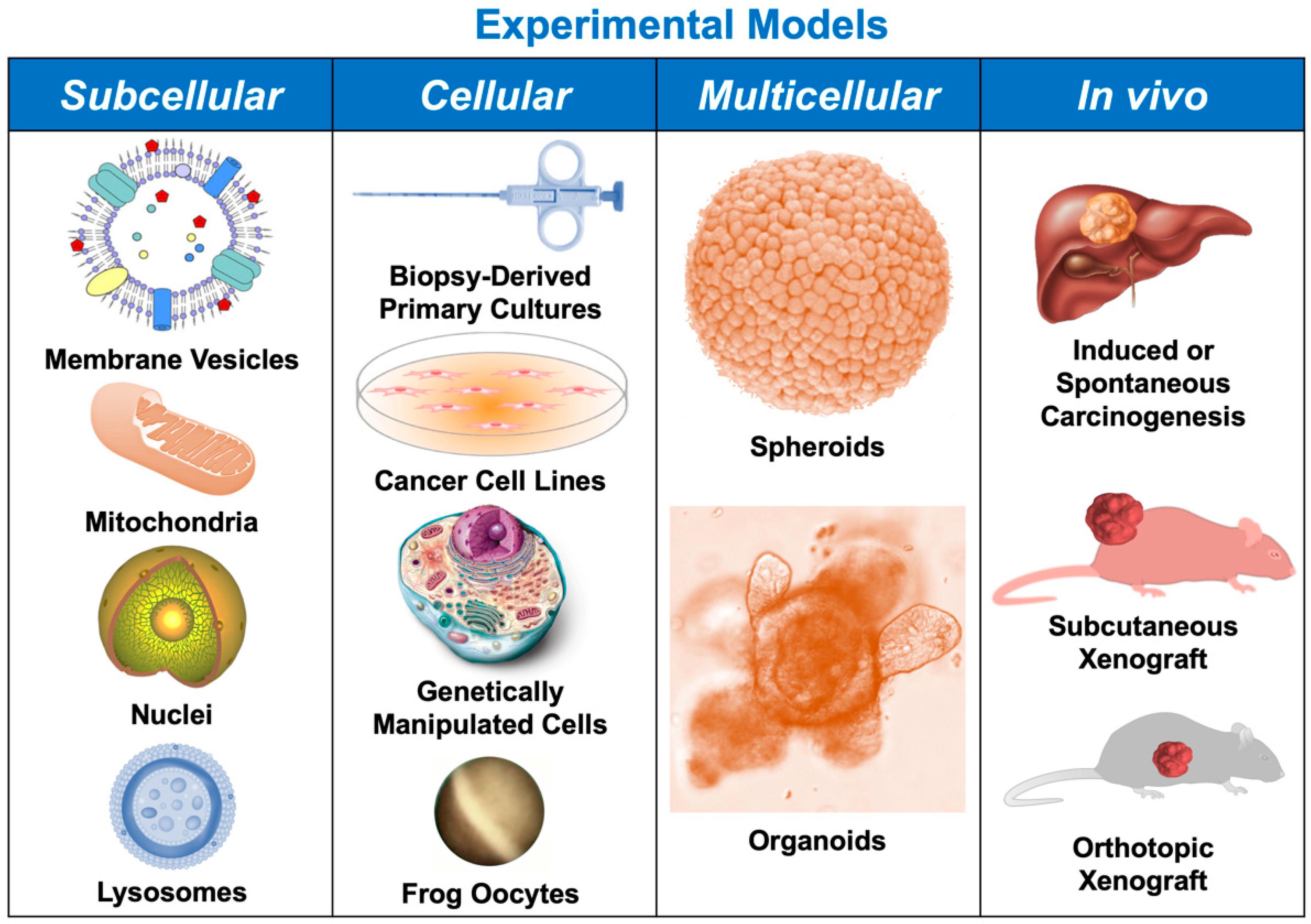
2. Subcellular Models
2.1. Plasma Membrane
2.2. Mitochondria
2.3. Nuclei
2.4. Lysosomes
3. Cellular Models
4. Spheroids and Organoids
5. In Vivo Models
5.1. HCC Models
5.2. CCA Models
5.3. HB Models
6. Conclusion and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
References
- Marin, J.J.G.; Briz, O.; Herraez, E.; Lozano, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Osorio-Padilla, L.M.; Santos-Llamas, A.I.; Serrano, M.A.; et al. Molecular bases of the poor response of liver cancer to chemotherapy. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Becerra, P.; Vaquero, J.; Romero, M.R.; Lozano, E.; Anadon, C.; Macias, R.I.; Serrano, M.A.; Grane-Boladeras, N.; Munoz-Bellvis, L.; Alvarez, L.; et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 2012, 9, 1693–1704. [Google Scholar] [CrossRef]
- Horio, M.; Gottesman, M.M.; Pastan, I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3580–3584. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.W.; Wring, S.A.; Humphreys, J.E.; Huang, L.; Morgan, J.B.; Webster, L.O.; Serabjit-Singh, C.S. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharm. Exp. Ther. 2001, 299, 620–628. [Google Scholar]
- Steck, T.L.; Weinstein, R.S.; Straus, J.H.; Wallach, D.F. Inside-out red cell membrane vesicles: preparation and purification. Science 1970, 168, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Mehn, D.; Jani, M.; Oosterhuis, B.; Heredi-Szabo, K.; Krajcsi, P. Utilization of membrane vesicle preparations to study drug-ABC transporter interactions. Expert Opin. Drug Metab. Toxic. 2008, 4, 721–732. [Google Scholar] [CrossRef]
- Aanismaa, P.; Seelig, A. P-Glycoprotein kinetics measured in plasma membrane vesicles and living cells. Biochemistry 2007, 46, 3394–3404. [Google Scholar] [CrossRef]
- Cui, Y.; Konig, J.; Buchholz, J.K.; Spring, H.; Leier, I.; Keppler, D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharm. 1999, 55, 929–937. [Google Scholar]
- Zeng, H.; Liu, G.; Rea, P.A.; Kruh, G.D. Transport of amphipathic anions by human multidrug resistance protein 3. Cancer Res. 2000, 60, 4779–4784. [Google Scholar]
- Wissel, G.; Deng, F.; Kudryavtsev, P.; Ghemtio, L.; Wipf, P.; Xhaard, H.; Kidron, H. A structure-activity relationship study of ABCC2 inhibitors. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2017, 103, 60–69. [Google Scholar] [CrossRef]
- Sjostedt, N.; Salminen, T.A.; Kidron, H. Endogenous, cholesterol-activated ATP-dependent transport in membrane vesicles from Spodoptera frugiperda cells. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 137, 104963. [Google Scholar] [CrossRef] [PubMed]
- Bravo, P.; Marin, J.J.; Beveridge, M.J.; Novak, D.A. Reconstitution and characterization of ATP-dependent bile acid transport in human and rat placenta. Biochem. J. 1995, 311 Pt. 2, 479–485. [Google Scholar] [CrossRef]
- Geertsma, E.R.; Nik Mahmood, N.A.; Schuurman-Wolters, G.K.; Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 2008, 3, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Price, E.M.; Boucher, R.C.; Germann, U.A.; Scarborough, G.A. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem. 1992, 267, 4854–4858. [Google Scholar] [PubMed]
- Javed, M.U.; Michelangeli, F. Enzymatic method for assaying calcium in serum with Ca++ -ATPase. Exp. Mol. Med. 2003, 35, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Brune, M.; Hunter, J.L.; Corrie, J.E.; Webb, M.R. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry 1994, 33, 8262–8271. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.; Nies, A.T.; Konig, J.; Frey, M.; Zentgraf, H.; Keppler, D. Purification of the human apical conjugate export pump MRP2 reconstitution and functional characterization as substrate-stimulated ATPase. Eur. J. Biochem. 1999, 265, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.G.; Karl, D.W. Inorganic phosphate assay with malachite green: An improvement and evaluation. J. Biochem. Biophys. Methods 1982, 7, 7–13. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Xiang, Q.F.; Zhang, D.M.; Wang, J.N.; Zhang, H.W.; Zheng, Z.Y.; Yu, D.C.; Li, Y.J.; Xu, J.; Chen, Y.J.; Shang, C.Z. Cabozantinib reverses multidrug resistance of human hepatoma HepG2/adr cells by modulating the function of P-glycoprotein. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1010–1023. [Google Scholar] [CrossRef]
- Li, X.Q.; Wang, L.; Lei, Y.; Hu, T.; Zhang, F.L.; Cho, C.H.; To, K.K. Reversal of P-gp and BCRP-mediated MDR by tariquidar derivatives. Eur. J. Med. Chem. 2015, 101, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Yamamoto, Y.; Taura, K.; Hata, K.; Fukumoto, M.; Uchinami, H.; Yonezawa, K.; Yamaoka, Y. Beneficial effect of cepharanthine on overcoming drug-resistance of hepatocellular carcinoma. Int. J. Oncol. 2004, 24, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.L.; Li, Y.J.; Yang, D.H.; Wang, C.R.; Xu, J.; Yao, N.; Zhang, X.Q.; Chen, Z.S.; Ye, W.C.; Zhang, D.M. Ganoderma lucidum derived ganoderenic acid B reverses ABCB1-mediated multidrug resistance in HepG2/ADM cells. Int. J. Oncol. 2015, 46, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Bohme, M.; Jedlitschky, G.; Leier, I.; Buchler, M.; Keppler, D. ATP-dependent export pumps and their inhibition by cyclosporins. Adv. Enzyme Regul. 1994, 34, 371–380. [Google Scholar] [CrossRef]
- Al-Shawi, M.K.; Polar, M.K.; Omote, H.; Figler, R.A. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 2003, 278, 52629–52640. [Google Scholar] [CrossRef] [PubMed]
- Urbatsch, I.L.; Sankaran, B.; Weber, J.; Senior, A.E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995, 270, 19383–19390. [Google Scholar] [CrossRef]
- Royo, F.; Falcon-Perez, J.M. Liver extracellular vesicles in health and disease. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef]
- Maacha, S.; Bhat, A.A.; Jimenez, L.; Raza, A.; Haris, M.; Uddin, S.; Grivel, J.C. Extracellular vesicles-mediated intercellular communication: Roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer 2019, 18, 55. [Google Scholar] [CrossRef]
- Wettmarshausen, J.; Perocchi, F. Isolation of Functional Mitochondria from Cultured Cells and Mouse Tissues. Methods Mol. Biol. 2017, 1567, 15–32. [Google Scholar] [CrossRef]
- Guo, W.; Dong, W.; Li, M.; Shen, Y. Mitochondria P-glycoprotein confers paclitaxel resistance on ovarian cancer cells. OncoTargets Ther. 2019, 12, 3881–3891. [Google Scholar] [CrossRef]
- Solazzo, M.; Fantappie, O.; Lasagna, N.; Sassoli, C.; Nosi, D.; Mazzanti, R. P-gp localization in mitochondria and its functional characterization in multiple drug-resistant cell lines. Exp. Cell Res. 2006, 312, 4070–4078. [Google Scholar] [CrossRef] [PubMed]
- Solazzo, M.; Fantappie, O.; D’Amico, M.; Sassoli, C.; Tani, A.; Cipriani, G.; Bogani, C.; Formigli, L.; Mazzanti, R. Mitochondrial expression and functional activity of breast cancer resistance protein in different multiple drug-resistant cell lines. Cancer Res. 2009, 69, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Baechler, S.A.; Factor, V.M.; Dalla Rosa, I.; Ravji, A.; Becker, D.; Khiati, S.; Miller Jenkins, L.M.; Lang, M.; Sourbier, C.; Michaels, S.A.; et al. The mitochondrial type IB topoisomerase drives mitochondrial translation and carcinogenesis. Nat. Commun. 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Wen, L.; Zhou, Y. Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. Int. J. Med. Sci. 2012, 9, 545–554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fantappie, O.; Sassoli, C.; Tani, A.; Nosi, D.; Marchetti, S.; Formigli, L.; Mazzanti, R. Mitochondria of a human multidrug-resistant hepatocellular carcinoma cell line constitutively express inducible nitric oxide synthase in the inner membrane. J. Cell. Mol. Med. 2015, 19, 1410–1417. [Google Scholar] [CrossRef] [PubMed]
- Serrano, F.A.; Matsuo, A.L.; Monteforte, P.T.; Bechara, A.; Smaili, S.S.; Santana, D.P.; Rodrigues, T.; Pereira, F.V.; Silva, L.S.; Machado, J., Jr.; et al. A cyclopalladated complex interacts with mitochondrial membrane thiol-groups and induces the apoptotic intrinsic pathway in murine and cisplatin-resistant human tumor cells. BMC Cancer 2011, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.; Rosales, R.; Macias, R.I.; Iannota, V.; Martinez-Fernandez, A.; Romero, M.R.; Hofmann, A.F.; Marin, J.J. Cytosol-nucleus traffic and colocalization with FXR of conjugated bile acids in rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G54–G62. [Google Scholar] [CrossRef]
- Rosales, R.; Monte, M.J.; Blazquez, A.G.; Briz, O.; Marin, J.J. ABCC2 is involved in the hepatocyte perinuclear barrier for small organic compounds. Biochem. Pharmacol. 2012, 84, 1651–1659. [Google Scholar] [CrossRef]
- Dudas, J.; Bocsi, J.; Fullar, A.; Baghy, K.; Fule, T.; Kudaibergenova, S.; Kovalszky, I. Heparin and liver heparan sulfate can rescue hepatoma cells from topotecan action. BioMed Res. Int. 2014, 2014, 765794. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Aguado, C.; Perez-Jimenez, E.; Lahuerta, M.; Knecht, E. Isolation of Lysosomes from Mammalian Tissues and Cultured Cells. Methods Mol. Biol. 2016, 1449, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Panse, M.; Radunski, U.; Koch, R.; Wenzel, D.; Inagaki, N.; Haase, D.; Truemper, L.; Wulf, G.G. ABC transporter A3 facilitates lysosomal sequestration of imatinib and modulates susceptibility of chronic myeloid leukemia cell lines to this drug. Haematologica 2009, 94, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 24, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 897–905. [Google Scholar] [CrossRef]
- Qiu, Z.; Zou, K.; Zhuang, L.; Qin, J.; Li, H.; Li, C.; Zhang, Z.; Chen, X.; Cen, J.; Meng, Z.; et al. Hepatocellular carcinoma cell lines retain the genomic and transcriptomic landscapes of primary human cancers. Sci. Rep. 2016, 6, 27411. [Google Scholar] [CrossRef]
- Rikhi, R.R.; Spady, K.K.; Hoffman, R.I.; Bateman, M.S.; Bateman, M.; Howard, L.E. Hepatoblastoma: A Need for Cell Lines and Tissue Banks to Develop Targeted Drug Therapies. Front. Pediatr. 2016, 4, 22. [Google Scholar] [CrossRef]
- Zach, S.; Birgin, E.; Rückert, F. Primary Cholangiocellular Carcinoma Cell Lines. J. Stem Cell Res. Transplant. 2015, 2, 10–13. [Google Scholar]
- Jouan, E.; Le Vee, M.; Denizot, C.; Parmentier, Y.; Fardel, O. Drug Transporter Expression and Activity in Human Hepatoma HuH-7 Cells. Pharmaceutics 2016, 9, 3. [Google Scholar] [CrossRef]
- Guttmann, S.; Chandhok, G.; Groba, S.R.; Niemietz, C.; Sauer, V.; Gomes, A.; Ciarimboli, G.; Karst, U.; Zibert, A.; Schmidt, H.H. Organic cation transporter 3 mediates cisplatin and copper cross-resistance in hepatoma cells. Oncotarget 2018, 9, 743–754. [Google Scholar] [CrossRef]
- Rodrigues, A.C.; Curi, R.; Genvigir, F.D.; Hirata, M.H.; Hirata, R.D. The expression of efflux and uptake transporters are regulated by statins in Caco-2 and HepG2 cells. Acta Pharmacologica Sinica 2009, 30, 956–964. [Google Scholar] [CrossRef]
- Herraez, E.; Lozano, E.; Macias, R.I.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Asensio, M.; Lozano, E.; Cives-Losada, C.; Carrillo, J.; Abete, L.; Briz, O.; Al-Abdulla, R.; Alonso-Peña, M.; Perez-Silva, L.; Cairo, S.; et al. Role of transportome in chemoresistance of hepatoblastoma. J. Physiol. Biochem. 2018, 74, S90. [Google Scholar]
- Chen, Y.B.; Yan, M.L.; Gong, J.P.; Xia, R.P.; Liu, L.X.; Li, N.; Lu, S.C.; Zhang, J.G.; Zeng, D.B.; Xie, J.G.; et al. Establishment of hepatocellular carcinoma multidrug resistant monoclone cell line HepG2/mdr1. Ch. Med. J. 2007, 120, 703–707. [Google Scholar] [CrossRef]
- Takahata, T.; Ookawa, K.; Suto, K.; Tanaka, M.; Yano, H.; Nakashima, O.; Kojiro, M.; Tamura, Y.; Tateishi, T.; Sakata, Y.; et al. Chemosensitivity determinants of irinotecan hydrochloride in hepatocellular carcinoma cell lines. Basic Clin. Pharmacol. Toxicol. 2008, 102, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.; Nakahashi, Y.; Hachimine, D.; Seki, T.; Okazaki, K. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins. Int. J. Oncol. 2007, 31, 1465–1472. [Google Scholar] [CrossRef]
- Tepsiri, N.; Chaturat, L.; Sripa, B.; Namwat, W.; Wongkham, S.; Bhudhisawasdi, V.; Tassaneeyakul, W. Drug sensitivity and drug resistance profiles of human intrahepatic cholangiocarcinoma cell lines. World J. Gastroenterol. 2005, 11, 2748–2753. [Google Scholar] [CrossRef]
- Hu, D.G.; Mackenzie, P.I.; Lu, L.; Meech, R.; McKinnon, R.A. Induction of human UDP-Glucuronosyltransferase 2B7 gene expression by cytotoxic anticancer drugs in liver cancer HepG2 cells. Drug Metab. Dispos. Biol. Fate Chem. 2015, 43, 660–668. [Google Scholar] [CrossRef]
- Zeekpudsa, P.; Kukongviriyapan, V.; Senggunprai, L.; Sripa, B.; Prawan, A. Suppression of NAD(P)H-quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J. Exp. Clin. Cancer Res. CR 2014, 33, 11. [Google Scholar] [CrossRef]
- Nakajima, T.; Takayama, T.; Miyanishi, K.; Nobuoka, A.; Hayashi, T.; Abe, T.; Kato, J.; Sakon, K.; Naniwa, Y.; Tanabe, H.; et al. Reversal of multiple drug resistance in cholangiocarcinoma by the glutathione S-transferase-pi-specific inhibitor O1-hexadecyl-gamma-glutamyl-S-benzylcysteinyl-D-phenylglycine ethylester. J. Pharmacol. Exp. Ther. 2003, 306, 861–869. [Google Scholar] [CrossRef]
- Dumontet, C.; Sikic, B.I. Mechanisms of action of and resistance to antitubulin agents: Microtubule dynamics, drug transport, and cell death. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 1061–1070. [Google Scholar] [CrossRef]
- Pang, E.; Hu, Y.; Chan, K.Y.; Lai, P.B.; Squire, J.A.; Macgregor, P.F.; Beheshti, B.; Albert, M.; Leung, T.W.; Wong, N. Karyotypic imbalances and differential gene expressions in the acquired doxorubicin resistance of hepatocellular carcinoma cells. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 664–674. [Google Scholar] [CrossRef]
- Chan, K.T.; Lung, M.L. Mutant p53 expression enhances drug resistance in a hepatocellular carcinoma cell line. Cancer Chemother. Pharmacol. 2004, 53, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Lin, Z.B.; Tan, H.R. Wild type p53 increased chemosensitivity of drug-resistant human hepatocellular carcinoma Bel7402/5-FU cells. Acta Pharmacologica Sinica 2004, 25, 76–82. [Google Scholar]
- Takeba, Y.; Sekine, S.; Kumai, T.; Matsumoto, N.; Nakaya, S.; Tsuzuki, Y.; Yanagida, Y.; Nakano, H.; Asakura, T.; Ohtsubo, T.; et al. Irinotecan-induced apoptosis is inhibited by increased P-glycoprotein expression and decreased p53 in human hepatocellular carcinoma cells. Biol. Pharm. Bull. 2007, 30, 1400–1406. [Google Scholar] [CrossRef]
- Namwat, N.; Amimanan, P.; Loilome, W.; Jearanaikoon, P.; Sripa, B.; Bhudhisawasdi, V.; Tassaneeyakul, W. Characterization of 5-fluorouracil-resistant cholangiocarcinoma cell lines. Chemotherapy 2008, 54, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Chun, E.; Lee, K.Y. Bcl-2 and Bcl-xL are important for the induction of paclitaxel resistance in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2004, 315, 771–779. [Google Scholar] [CrossRef]
- Yoon, H.; Min, J.K.; Lee, J.W.; Kim, D.G.; Hong, H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem. Biophys. Res. Commun. 2011, 405, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Wattanawongdon, W.; Hahnvajanawong, C.; Namwat, N.; Kanchanawat, S.; Boonmars, T.; Jearanaikoon, P.; Leelayuwat, C.; Techasen, A.; Seubwai, W. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int. J. Oncol. 2015, 47, 398–410. [Google Scholar] [CrossRef]
- Fleischer, B.; Schulze-Bergkamen, H.; Schuchmann, M.; Weber, A.; Biesterfeld, S.; Muller, M.; Krammer, P.H.; Galle, P.R. Mcl-1 is an anti-apoptotic factor for human hepatocellular carcinoma. Int. J. Oncol. 2006, 28, 25–32. [Google Scholar] [CrossRef]
- Na, D.C.; Lee, J.E.; Yoo, J.E.; Oh, B.K.; Choi, G.H.; Park, Y.N. Invasion and EMT-associated genes are up-regulated in B viral hepatocellular carcinoma with high expression of CD133-human and cell culture study. Exp. Mol. Pathol. 2011, 90, 66–73. [Google Scholar] [CrossRef]
- Cheung, P.F.; Yip, C.W.; Ng, L.W.; Lo, K.W.; Wong, N.; Choy, K.W.; Chow, C.; Chan, K.F.; Cheung, T.T.; Poon, R.T.; et al. Establishment and characterization of a novel primary hepatocellular carcinoma cell line with metastatic ability in vivo. Cancer Cell Int. 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Y.; Chuang, W.L. Genes responsible for the characteristics of primary cultured invasive phenotype hepatocellular carcinoma cells. Biomed. Pharmacother. Biomed. Pharmacotherapie 2012, 66, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Castven, D.; Becker, D.; Czauderna, C.; Wilhelm, D.; Andersen, J.B.; Strand, S.; Hartmann, M.; Heilmann-Heimbach, S.; Roth, W.; Hartmann, N.; et al. Application of patient-derived liver cancer cells for phenotypic characterization and therapeutic target identification. Int. J. Cancer 2019, 144, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Hong, Z.; Feng, F.; Chai, Y.; Zhang, Y.; Jiang, Q.; Hu, Y.; Wu, S.; Wu, Y.; Gao, X.; et al. A novel chemotherapeutic sensitivity-testing system based on collagen gel droplet embedded 3D-culture methods for hepatocellular carcinoma. BMC Cancer 2017, 17, 729. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Wu, C.C.; Chuang, Y.H.; Chuang, W.L. Clinical utility of a simple primary culture method in hepatocellular carcinoma patients. J. Gastroenterol. Hepatol. 2015, 30, 352–357. [Google Scholar] [CrossRef]
- Fraveto, A.; Cardinale, V.; Bragazzi, M.C.; Giuliante, F.; De Rose, A.M.; Grazi, G.L.; Napoletano, C.; Semeraro, R.; Lustri, A.M.; Costantini, D.; et al. Sensitivity of Human Intrahepatic Cholangiocarcinoma Subtypes to Chemotherapeutics and Molecular Targeted Agents: A Study on Primary Cell Cultures. PLoS ONE 2015, 10, e0142124. [Google Scholar] [CrossRef]
- Trumpp, A.; Wiestler, O.D. Mechanisms of Disease: Cancer stem cells—Targeting the evil twin. Nat. Clin. Pract. Oncol. 2008, 5, 337–347. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Sukowati, C.H.C. Heterogeneity of Hepatic Cancer Stem Cells. Adv. Exp. Med. Biol. 2019, 1139, 59–81. [Google Scholar] [CrossRef]
- Yamashita, T.; Wang, X.W. Cancer stem cells in the development of liver cancer. J. Clin. Investig. 2013, 123, 1911–1918. [Google Scholar] [CrossRef]
- Yuan, X.; Li, J.; Coulouarn, C.; Lin, T.; Sulpice, L.; Bergeat, D.; De La Torre, C.; Liebe, R.; Gretz, N.; Ebert, M.P.A.; et al. SOX9 expression decreases survival of patients with intrahepatic cholangiocarcinoma by conferring chemoresistance. Br. J. Cancer 2018, 119, 1358–1366. [Google Scholar] [CrossRef]
- Huang, L.; Cai, J.; Guo, H.; Gu, J.; Tong, Y.; Qiu, B.; Wang, C.; Li, M.; Xia, L.; Zhang, J.; et al. ID3 Promotes Stem Cell Features and Predicts Chemotherapeutic Response of Intrahepatic Cholangiocarcinoma. Hepatology 2019, 69, 1995–2012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Y.; Qiao, L.; Zhao, Y.; Wei, Y. Protective effects of hepatic stellate cells against cisplatin-induced apoptosis in human hepatoma G2 cells. Int. J. Oncol. 2015, 47, 632–640. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.; Gou, W.; Liu, M.; Sang, W.; Chu, H.; Zhang, W. Expression of P53 and HSP70 in Chronic Hepatitis, Liver Cirrhosis, and Early and Advanced Hepatocellular Carcinoma Tissues and Their Diagnostic Value in Hepatocellular Carcinoma: An Immunohistochemical Study. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, J.; Shi, H.; Wang, Z.; Zhang, G.; Cao, Y.; Jiang, C.; Ding, Y. Hepatic stellate cell coculture enables sorafenib resistance in Huh7 cells through HGF/c-Met/Akt and Jak2/Stat3 pathways. BioMed Res. Int. 2014, 2014, 764981. [Google Scholar] [CrossRef]
- Fujita, D.; Saito, Y.; Nakanishi, T.; Tamai, I. Organic Anion Transporting Polypeptide (OATP)2B1 Contributes to Gastrointestinal Toxicity of Anticancer Drug SN-38, Active Metabolite of Irinotecan Hydrochloride. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 1–7. [Google Scholar] [CrossRef]
- Nakanishi, T.; Doyle, L.A.; Hassel, B.; Wei, Y.; Bauer, K.S.; Wu, S.; Pumplin, D.W.; Fang, H.B.; Ross, D.D. Functional characterization of human breast cancer resistance protein (BCRP, ABCG2) expressed in the oocytes of Xenopus laevis. Mol. Pharmacol. 2003, 64, 1452–1462. [Google Scholar] [CrossRef]
- Janke, D.; Mehralivand, S.; Strand, D.; Godtel-Armbrust, U.; Habermeier, A.; Gradhand, U.; Fischer, C.; Toliat, M.R.; Fritz, P.; Zanger, U.M.; et al. 6-mercaptopurine and 9-(2-phosphonyl-methoxyethyl) adenine (PMEA) transport altered by two missense mutations in the drug transporter gene ABCC4. Hum. Mutat. 2008, 29, 659–669. [Google Scholar] [CrossRef]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, W.; Kang, L.; Wang, Y.; Zhang, M.; Zhang, H.; Hu, P. Microfluidic co-culture of liver tumor spheroids with stellate cells for the investigation of drug resistance and intercellular interactions. Analyst 2019, 144, 4233–4240. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarro, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef]
- Lampis, A.; Carotenuto, P.; Vlachogiannis, G.; Cascione, L.; Hedayat, S.; Burke, R.; Clarke, P.; Bosma, E.; Simbolo, M.; Scarpa, A.; et al. MIR21 Drives Resistance to Heat Shock Protein 90 Inhibition in Cholangiocarcinoma. Gastroenterology 2018, 154, 1066–1079. [Google Scholar] [CrossRef]
- Wang, Y.; Takeishi, K.; Li, Z.; Cervantes-Alvarez, E.; Collin de l’Hortet, A.; Guzman-Lepe, J.; Cui, X.; Zhu, J. Microenvironment of a tumor-organoid system enhances hepatocellular carcinoma malignancy-related hallmarks. Organogenesis 2017, 13, 83–94. [Google Scholar] [CrossRef]
- Nakagawa, H.; Suzuki, N.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Kinoshita, H.; Ihara, S.; Uchino, K.; Nishikawa, Y.; Ijichi, H.; et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl. Acad. Sci. USA 2017, 114, E3806–E3815. [Google Scholar] [CrossRef] [PubMed]
- Pitot, H.C.; Dragan, Y.P. Facts and theories concerning the mechanisms of carcinogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1991, 5, 2280–2286. [Google Scholar] [CrossRef]
- Frezza, E.E.; Gerunda, G.E.; Farinati, F.; DeMaria, N.; Galligioni, A.; Plebani, F.; Giacomin, A.; Van Thiel, D.H. CCL4-induced liver cirrhosis and hepatocellular carcinoma in rats: Relationship to plasma zinc, copper and estradiol levels. Hepato-Gastroenterol. 1994, 41, 367–369. [Google Scholar]
- Shiota, G.; Harada, K.; Ishida, M.; Tomie, Y.; Okubo, M.; Katayama, S.; Ito, H.; Kawasaki, H. Inhibition of hepatocellular carcinoma by glycyrrhizin in diethylnitrosamine-treated mice. Carcinogenesis 1999, 20, 59–63. [Google Scholar] [CrossRef]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Casadei Gardini, A.; Marisi, G.; Baron Toaldo, M.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef]
- Farber, E.; Solt, D.; Cameron, R.; Laishes, B.; Ogawa, K.; Medline, A. Newer insights into the pathogenesis of liver cancer. Am. J. Pathol. 1977, 89, 477–482. [Google Scholar]
- Al-Abdulla, R.; Lozano, E.; Macias, R.I.R.; Monte, M.J.; Briz, O.; O’Rourke, C.J.; Serrano, M.A.; Banales, J.M.; Avila, M.A.; Martinez-Chantar, M.L.; et al. Epigenetic events involved in organic cation transporter 1-dependent impaired response of hepatocellular carcinoma to sorafenib. Br. J. Pharmacol. 2019, 176, 787–800. [Google Scholar] [CrossRef]
- Hays, T.; Rusyn, I.; Burns, A.M.; Kennett, M.J.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 2005, 26, 219–227. [Google Scholar] [CrossRef]
- Woo, L.L.; Egner, P.A.; Belanger, C.L.; Wattanawaraporn, R.; Trudel, L.J.; Croy, R.G.; Groopman, J.D.; Essigmann, J.M.; Wogan, G.N.; Bouhenguel, J.T. Aflatoxin B1-DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxic. Sci. Off. J. Soc. Toxicol. 2011, 122, 38–44. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Hunter, K.; LeVoyer, T.; Roush, J.; Wise, P.; Michielli, R.A.; Shen, F.M.; Evans, A.A.; London, W.T.; Buetow, K.H. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res. 2003, 63, 4594–4601. [Google Scholar]
- Knight, B.; Yeoh, G.C.; Husk, K.L.; Ly, T.; Abraham, L.J.; Yu, C.; Rhim, J.A.; Fausto, N. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J. Exp. Med. 2000, 192, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T., Jr.; Steelman, L.S.; McCubrey, J.A. Phosphatidylinositol 3′-kinase activation leads to multidrug resistance protein-1 expression and subsequent chemoresistance in advanced prostate cancer cells. Cancer Res. 2004, 64, 8397–8404. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Dong, X.; Zhai, B.; Jiang, X.; Dong, D.; Li, B.; Jiang, H.; Xu, S.; Sun, X. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 2015, 6, 28867–28881. [Google Scholar] [CrossRef]
- Tang, T.C.; Man, S.; Xu, P.; Francia, G.; Hashimoto, K.; Emmenegger, U.; Kerbel, R.S. Development of a resistance-like phenotype to sorafenib by human hepatocellular carcinoma cells is reversible and can be delayed by metronomic UFT chemotherapy. Neoplasia 2010, 12, 928–940. [Google Scholar] [CrossRef]
- Briz, O.; Macias, R.I.; Vallejo, M.; Silva, A.; Serrano, M.A.; Marin, J.J. Usefulness of liposomes loaded with cytostatic bile acid derivatives to circumvent chemotherapy resistance of enterohepatic tumors. Mol. Pharmacol. 2003, 63, 742–750. [Google Scholar] [CrossRef]
- Kornek, M.; Raskopf, E.; Tolba, R.; Becker, U.; Klockner, M.; Sauerbruch, T.; Schmitz, V. Accelerated orthotopic hepatocellular carcinomas growth is linked to increased expression of pro-angiogenic and prometastatic factors in murine liver fibrosis. Liver Int. Off. J. Int. Assoc. Study Liver 2008, 28, 509–518. [Google Scholar] [CrossRef]
- Hollingshead, M.G.; Alley, M.C.; Camalier, R.F.; Abbott, B.J.; Mayo, J.G.; Malspeis, L.; Grever, M.R. In vivo cultivation of tumor cells in hollow fibers. Life Sci. 1995, 57, 131–141. [Google Scholar] [CrossRef]
- Urbanik, T.; Boger, R.J.; Longerich, T.; Becker, K.; Ehrenberg, K.R.; Hovelmeyer, N.; Hahn, M.; Schuchmann, M.; Jager, D.; Waisman, A.; et al. Liver specific deletion of CYLDexon7/8 induces severe biliary damage, fibrosis and increases hepatocarcinogenesis in mice. J. Hepatol. 2012, 57, 995–1003. [Google Scholar] [CrossRef]
- Praet, M.M.; Roels, H.J. Histogenesis of cholangiomas and cholangiocarcinomas in thioacetamide fed rats. Exp. Pathol. 1984, 26, 3–14. [Google Scholar] [CrossRef]
- Mansuroglu, T.; Ramadori, P.; Dudas, J.; Malik, I.; Hammerich, K.; Fuzesi, L.; Ramadori, G. Expression of stem cell factor and its receptor c-Kit during the development of intrahepatic cholangiocarcinoma. Lab. Investig. J. Tech. Methods Pathol. 2009, 89, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.M.; Cheng, C.T.; Wu, R.C.; Chung, Y.H.; Chiang, K.C.; Yeh, T.S.; Liu, C.Y.; Chen, M.H.; Yeh, C.N. Nab-paclitaxel is effective against intrahepatic cholangiocarcinoma via disruption of desmoplastic stroma. Oncol. Lett. 2018, 16, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Sanchez-Vicente, L.; Monte, M.J.; Herraez, E.; Briz, O.; Banales, J.M.; Marin, J.J.; Macias, R.I. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 2014, 12, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Hylemon, P.B.; Zhou, H. Conjugated Bile Acids Promote Invasive Growth of Esophageal Adenocarcinoma Cells and Cancer Stem Cell Expansion via Sphingosine 1-Phosphate Receptor 2-Mediated Yes-Associated Protein Activation. Am. J. Pathol. 2018, 188, 2042–2058. [Google Scholar] [CrossRef]
- O’Dell, M.R.; Huang, J.L.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef]
- Vasey, P.A.; Jones, N.A.; Jenkins, S.; Dive, C.; Brown, R. Cisplatin, camptothecin, and taxol sensitivities of cells with p53-associated multidrug resistance. Mol. Pharm. 1996, 50, 1536–1540. [Google Scholar]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef]
- Yokoyama, M.; Ohnishi, H.; Ohtsuka, K.; Matsushima, S.; Ohkura, Y.; Furuse, J.; Watanabe, T.; Mori, T.; Sugiyama, M. KRAS Mutation as a Potential Prognostic Biomarker of Biliary Tract Cancers. Jpn. Clin. Med. 2016, 7, 33–39. [Google Scholar] [CrossRef]
- Urtasun, N.; Boces-Pascual, C.; Boix, L.; Bruix, J.; Pastor-Anglada, M.; Perez-Torras, S. Role of drug-dependent transporter modulation on the chemosensitivity of cholangiocarcinoma. Oncotarget 2017, 8, 90185–90196. [Google Scholar] [CrossRef]
- Ren, H.Y.; Chen, B.; Huang, G.L.; Liu, Y.; Shen, D.Y. Upregulation of retinoic acid receptor-beta reverses drug resistance in cholangiocarcinoma cells by enhancing susceptibility to apoptosis. Mol. Med. Rep. 2016, 14, 3602–3608. [Google Scholar] [CrossRef]
- Hong, Z.F.; Zhao, W.X.; Yin, Z.Y.; Xie, C.R.; Xu, Y.P.; Chi, X.Q.; Zhang, S.; Wang, X.M. Capsaicin Enhances the Drug Sensitivity of Cholangiocarcinoma through the Inhibition of Chemotherapeutic-Induced Autophagy. PLoS ONE 2015, 10, e0121538. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Lin, H.Y.; Chang, V.H.; Chen, C.C.; Liu, Y.R.; Wang, J.; Zhang, K.; Jiang, X.; Yen, Y. Lovastatin overcomes gefitinib resistance through TNF-alpha signaling in human cholangiocarcinomas with different LKB1 statuses in vitro and in vivo. Oncotarget 2015, 6, 23857–23873. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Macias, R.I.R.; Monte, M.J.; Asensio, M.; Del Carmen, S.; Sanchez-Vicente, L.; Alonso-Pena, M.; Al-Abdulla, R.; Munoz-Garrido, P.; Satriano, L.; et al. Causes of hOCT1-Dependent Cholangiocarcinoma Resistance to Sorafenib and Sensitization by Tumor-Selective Gene Therapy. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Yokomuro, S.; Tsuji, H.; Lunz, J.G., 3rd; Sakamoto, T.; Ezure, T.; Murase, N.; Demetris, A.J. Growth control of human biliary epithelial cells by interleukin 6, hepatocyte growth factor, transforming growth factor beta1, and activin A: comparison of a cholangiocarcinoma cell line with primary cultures of non-neoplastic biliary epithelial cells. Hepatology 2000, 32, 26–35. [Google Scholar] [CrossRef]
- Cadamuro, M.; Brivio, S.; Mertens, J.; Vismara, M.; Moncsek, A.; Milani, C.; Fingas, C.; Cristina Malerba, M.; Nardo, G.; Dall’Olmo, L.; et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol. 2019, 70, 700–709. [Google Scholar] [CrossRef]
- Fabris, L.; Cadamuro, M.; Moserle, L.; Dziura, J.; Cong, X.; Sambado, L.; Nardo, G.; Sonzogni, A.; Colledan, M.; Furlanetto, A.; et al. Nuclear expression of S100A4 calcium-binding protein increases cholangiocarcinoma invasiveness and metastasization. Hepatology 2011, 54, 890–899. [Google Scholar] [CrossRef]
- Sirica, A.E.; Zhang, Z.; Lai, G.H.; Asano, T.; Shen, X.N.; Ward, D.J.; Mahatme, A.; Dewitt, J.L. A novel “patient-like” model of cholangiocarcinoma progression based on bile duct inoculation of tumorigenic rat cholangiocyte cell lines. Hepatology 2008, 47, 1178–1190. [Google Scholar] [CrossRef]
- Nicolle, D.; Fabre, M.; Simon-Coma, M.; Gorse, A.; Kappler, R.; Nonell, L.; Mallo, M.; Haidar, H.; Deas, O.; Mussini, C.; et al. Patient-derived mouse xenografts from pediatric liver cancer predict tumor recurrence and advise clinical management. Hepatology 2016, 64, 1121–1135. [Google Scholar] [CrossRef]
- Warmann, S.; Hunger, M.; Teichmann, B.; Flemming, P.; Gratz, K.F.; Fuchs, J. The role of the MDR1 gene in the development of multidrug resistance in human hepatoblastoma: Clinical course and in vivo model. Cancer 2002, 95, 1795–1801. [Google Scholar] [CrossRef]
- Warmann, S.W.; Heitmann, H.; Teichmann, B.; Gratz, K.F.; Ruck, P.; Hunger, M.; Fuchs, J. Effects of P-glycoprotein modulation on the chemotherapy of xenotransplanted human hepatoblastoma. Pediatr. Hematol. Oncol. 2005, 22, 373–386. [Google Scholar] [CrossRef]
- Lieber, J.; Ellerkamp, V.; Vogt, F.; Wenz, J.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. BH3-mimetic drugs prevent tumour onset in an orthotopic mouse model of hepatoblastoma. Exp. Cell Res. 2014, 322, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [PubMed]
- Ertel, A.; Verghese, A.; Byers, S.W.; Ochs, M.; Tozeren, A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol. Cancer 2006, 5, 55. [Google Scholar] [CrossRef]


{kind=link}
| Model | Advantages | Limitations | Usefulness in Chemoresistance Research |
|---|---|---|---|
| Carcinogen-induced tumors | Similarity with the carcinogenesis phases seen in humans | Time consuming high number of animals needed | Intrinsic resistance study of changes in gene expression during carcinogenesis |
| Genetically engineered mouse models | Facilitate detailed investigation of carcinogenic pathways | Complex and time-consuming breeding strategies | Study of the role of oncogenes and tumor suppressor genes in MDR |
| Ectopic implants | Fast tumor development Easy to perform No post-operative complications | Important differences between cell lines No interaction with liver tissue Model of advanced tumor stage Absence of metastasis | Drug screening in resistant cells Evaluation of the role of MOC genes using modified cell lines |
| Orthotopic implants | Fast tumor development allows complex tumor-host interactions to be tested Feasibility to use fibrotic liver | Difficult procedure Important differences between cell lines The need of immunodeficient animals makes less realistic the tumor microenvironment | Drug screening in resistant cells Tumor-host interaction Evaluation of the role of MOC genes using modified cell lines |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, J.J.G.; Herraez, E.; Lozano, E.; Macias, R.I.R.; Briz, O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers 2019, 11, 1677. https://doi.org/10.3390/cancers11111677
Marin JJG, Herraez E, Lozano E, Macias RIR, Briz O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers. 2019; 11(11):1677. https://doi.org/10.3390/cancers11111677
Chicago/Turabian StyleMarin, Jose J. G., Elisa Herraez, Elisa Lozano, Rocio I. R. Macias, and Oscar Briz. 2019. "Models for Understanding Resistance to Chemotherapy in Liver Cancer" Cancers 11, no. 11: 1677. https://doi.org/10.3390/cancers11111677
APA StyleMarin, J. J. G., Herraez, E., Lozano, E., Macias, R. I. R., & Briz, O. (2019). Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers, 11(11), 1677. https://doi.org/10.3390/cancers11111677