Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
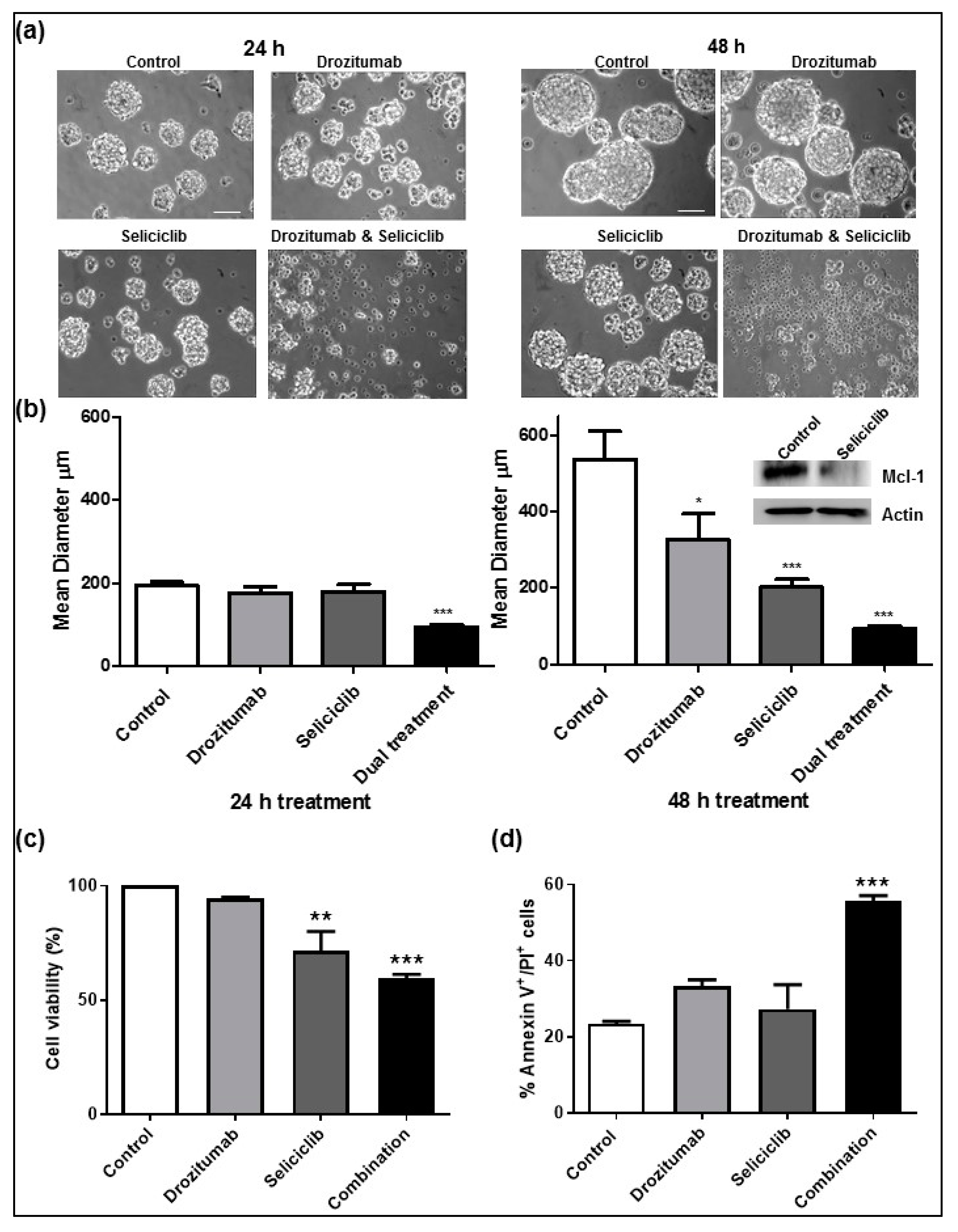
2.1. Seliciclib When Combined with Drozitumab, Reduces Both the Diameter and Viability of Human GBM Neurospheres and Induces Apoptosis In Vitro
2.2. In Vivo Toxicity Findings Associated with Seliciclib Plus Drozitumab Combinatorial Regimen
2.3. In Vivo Efficacy of Seliciclib Plus Drozitumab Combined Treatment in an Orthotopic PDX Model
2.4. Pharmacodynamics of Second-Generation CDK Inhibitor CYC065, In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Human GBM Neurosphere Culture
4.3. Light Microscopy
4.4. Cell Viability Assay
4.5. Flow Cytometry
4.6. Toxicity Assessment of Seliciclib Plus Drozitumab Combined Treatment in Rodent Models
4.7. Assessment of Seliciclib Plus Drozitumab Combinatorial Regimen in a PDX Model
4.8. BLI
4.9. Pharmacodynamic Analysis of Second-Generation CDK Inhibitor CYC065, in a GBM Orthotopic PDX Model
4.10. Western Blot Analysis
4.11. Histological Assessment and Immunohistochemistry
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McNamara, M.G.; Lwin, Z.; Jiang, H.; Chung, C.; Millar, B.A.; Sahgal, A.; Laperriere, N.; Mason, W.P. Conditional probability of survival and post-progression survival in patients with glioblastoma in the temozolomide treatment era. J. Neuro Oncol. 2014, 117, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Vymazal, J.; Wong, E.T. Response patterns of recurrent glioblastomas treated with tumor-treating fields. Semin. Oncol. 2014, 41, S14–S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Weyhenmeyer, B.; Murphy, A.C.; Prehn, J.H.; Murphy, B.M. Targeting the anti-apoptotic Bcl-2 family members for the treatment of cancer. Exp. Oncol. 2012, 34, 192–199. [Google Scholar]
- Murphy, B.M.; O’Neill, A.J.; Adrain, C.; Watson, R.W.; Martin, S.J. The apoptosome pathway to caspase activation in primary human neutrophils exhibits dramatically reduced requirements for cytochrome C. J. Exp. Med. 2003, 197, 625–632. [Google Scholar] [CrossRef]
- Adrain, C.; Murphy, B.M.; Martin, S.J. Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J. Biol. Chem. 2005, 280, 4663–4673. [Google Scholar] [CrossRef] [Green Version]
- Murphy, B.M.; Creagh, E.M.; Martin, S.J. Interchain proteolysis, in the absence of a dimerization stimulus, can initiate apoptosis-associated caspase-8 activation. J. Biol. Chem. 2004, 279, 36916–36922. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Michod, D.; Walicki, J.; Murphy, B.M.; Kasibhatla, S.; Martin, S.J.; Widmann, C. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol. Cell Biol. 2004, 24, 10425–10436. [Google Scholar] [CrossRef] [Green Version]
- Strasser, A.; Harris, A.W.; Huang, D.C.; Krammer, P.H.; Cory, S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995, 14, 6136–6147. [Google Scholar] [CrossRef]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.C.; Weyhenmeyer, B.; Schmid, J.; Kilbride, S.M.; Rehm, M.; Huber, H.J.; Senft, C.; Weissenberger, J.; Seifert, V.; Dunst, M.; et al. Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: A systems medicine approach. Cell Death Dis. 2013, 4, e629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 92, 57–70. [Google Scholar] [CrossRef]
- Strik, H.; Deininger, M.; Streffer, J.; Grote, E.; Wickboldt, J.; Dichgans, J.; Weller, M.; Meyermann, R. BCL-2 family protein expression in initial and recurrent glioblastomas: Modulation by radiochemotherapy. J. Neurol. Neurosurg. Psychiatry 1999, 67, 763–768. [Google Scholar] [CrossRef]
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Targeting the extrinsic apoptotic pathway in cancer: Lessons learned and future directions. J. Clin. Investig. 2015, 125, 487–489. [Google Scholar] [CrossRef]
- Kuijlen, J.M.; Bremer, E.; Mooij, J.J.; den Dunnen, W.F.; Helfrich, W. Review: On TRAIL for malignant glioma therapy? Neuropathol. Appl. Neurobiol. 2010, 36, 168–182. [Google Scholar] [CrossRef]
- Hetschko, H.; Voss, V.; Horn, S.; Seifert, V.; Prehn, J.H.; Kogel, D. Pharmacological inhibition of Bcl-2 family members reactivates TRAIL-induced apoptosis in malignant glioma. J. Neuro Oncol. 2008, 86, 265–272. [Google Scholar] [CrossRef]
- Fulda, S.; Wick, W.; Weller, M.; Debatin, K.M. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat. Med. 2002, 8, 808–815. [Google Scholar] [CrossRef]
- Murphy, A.C.; Weyhenmeyer, B.; Noonan, J.; Kilbride, S.M.; Schimansky, S.; Loh, K.P.; Kogel, D.; Letai, A.G.; Prehn, J.H.; Murphy, B.M. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis 2014, 19, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Nurse, P.; Masui, Y.; Hartwell, L. Understanding the cell cycle. Nat. Med. 1998, 4, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Hunt, T.; Nasmyth, K.; Novak, B. The cell cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011, 366, 3494–3497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, D.A.; Kim, J.S.; Jenkins, S.; Ressom, H.; Huang, M.; Coppa, N.; Mabanta, L.; Bigner, D.; Yan, H.; Jean, W.; et al. Identification of p18 INK4c as a tumor suppressor gene in glioblastoma multiforme. Cancer Res. 2008, 68, 2564–2569. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Turner, N.C.; Huang Bartlett, C.; Cristofanilli, M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 1672–1673. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Schroder, L.B.; McDonald, K.L. CDK4/6 Inhibitor PD0332991 in Glioblastoma Treatment: Does It Have a Future? Front. Oncol. 2015, 5, 259. [Google Scholar] [CrossRef]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef]
- Cocco, E.; Lopez, S.; Black, J.; Bellone, S.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; Schwab, C.L.; Menderes, G.; Zammataro, L.; et al. Dual CCNE1/PIK3CA targeting is synergistic in CCNE1-amplified/PIK3CA-mutated uterine serous carcinomas in vitro and in vivo. Br. J. Cancer 2016, 115, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.; Totpal, K.; Lawrence, D.; Marsters, S.; Pitti, R.; Yee, S.; Ross, S.; Deforge, L.; Koeppen, H.; Sagolla, M.; et al. Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ. 2008, 15, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Cristofanon, S.; Abhari, B.A.; Krueger, M.; Tchoghandjian, A.; Momma, S.; Calaminus, C.; Vucic, D.; Pichler, B.J.; Fulda, S. Identification of RIP1 as a critical mediator of Smac mimetic-mediated sensitization of glioblastoma cells for Drozitumab-induced apoptosis. Cell Death Dis. 2015, 6, e1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, A.T.; Alferez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinska, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 632. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Z.; Chen, J.J.; Yu, Y.; Li, B.; Sun, S.Y.; Zhang, B.; Cao, L. Drozitumab, a human antibody to death receptor 5, has potent antitumor activity against rhabdomyosarcoma with the expression of caspase-8 predictive of response. Clin. Cancer Res. 2011, 17, 3181–3192. [Google Scholar] [CrossRef] [Green Version]
- McClue, S.J.; Blake, D.; Clarke, R.; Cowan, A.; Cummings, L.; Fischer, P.M.; MacKenzie, M.; Melville, J.; Stewart, K.; Wang, S.; et al. In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int. J. Cancer 2002, 102, 463–468. [Google Scholar] [CrossRef]
- Vita, M.; Abdel-Rehim, M.; Olofsson, S.; Hassan, Z.; Meurling, L.; Siden, A.; Siden, M.; Pettersson, T.; Hassan, M. Tissue distribution, pharmacokinetics and identification of roscovitine metabolites in rat. Eur. J. Pharm. Sci. 2005, 25, 91–103. [Google Scholar] [CrossRef]
- Blake, D.G.; MacKay, C.; Frame, S.; Zheleva, D. CYC065, a novel CDK2/9 inhibitor: Molecular basis for clinical development in basal-like triple-negative breast cancer. In Proceedings of the Thirty-Eighth Annual CTRC-AACR San Antonio Breast Cancer Symposium, San Antonio, TX, USA, 8–12 December 2015. [Google Scholar]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Mallinger, A.; Workman, P.; Clarke, P.A. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol. Ther. 2017, 173, 83–105. [Google Scholar] [CrossRef]
- Hayashi, T.; Adachi, K.; Ohba, S.; Hirose, Y. The Cdk inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells. J. Neurooncol. 2013, 115, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, S.U.; Shin, D.Y.; Choi, K.S. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 2004, 23, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yushan, R.; Wenjie, C.; Suning, H.; Yiwu, D.; Tengfei, Z.; Madushi, W.M.; Feifei, L.; Changwen, Z.; Xin, W.; Roodrajeetsing, G.; et al. Insights into the clinical value of cyclin-dependent kinase 5 in glioma: A retrospective study. World J. Surg. Oncol. 2015, 13, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.; Meyer, D.; Guo, A.; Gundara, J.; Mettlach, G.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Tian, B.; Gearing, M.; Hunter, S.; Ye, K.; Mao, Z. Cdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 7570–7575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Tucker-Burden, C.; Kaissi, E.; Newsam, A.; Duggireddy, H.; Chau, M.; Zhang, C.; Diwedi, B.; Rupji, M.; Seby, S.; et al. CDK5 Inhibition Resolves PKA/cAMP-Independent Activation of CREB1 Signaling in Glioma Stem Cells. Cell Rep. 2018, 23, 1651–1664. [Google Scholar] [CrossRef] [Green Version]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef] [Green Version]
- Stuckey, D.W.; Shah, K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, J.; Jamoona, A.; Gulati, N.D.; Mohan, A.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Revisiting the role of p53 in primary and secondary glioblastomas. Anticancer Res. 2006, 26, 4633–4639. [Google Scholar] [PubMed]
- Chiang, M.F.; Chou, P.Y.; Wang, W.J.; Sze, C.I.; Chang, N.S. Tumor Suppressor WWOX and p53 Alterations and Drug Resistance in Glioblastomas. Front. Oncol. 2013, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, H.S.; Mitev, V.; Vlaykova, T.; Cavicchi, L.; Zhelev, N. Discovery and development of Seliciclib. How systems biology approaches can lead to better drug performance. J. Biotechnol. 2015, 202, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Mehta, S.; Huillard, E.; Kesari, S.; Maire, C.L.; Golebiowski, D.; Harrington, E.P.; Alberta, J.A.; Kane, M.F.; Theisen, M.; Ligon, K.L.; et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell 2011, 19, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef]
- Rubin, J.B.; Kung, A.L.; Klein, R.S.; Chan, J.A.; Sun, Y.; Schmidt, K.; Kieran, M.W.; Luster, A.D.; Segal, R.A. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 13513–13518. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Jarzabek, M.A.; Huszthy, P.C.; Skaftnesmo, K.O.; McCormack, E.; Dicker, P.; Prehn, J.H.; Bjerkvig, R.; Byrne, A.T. In vivo bioluminescence imaging validation of a human biopsy-derived orthotopic mouse model of glioblastoma multiforme. Mol. Imaging 2013, 12, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Filbin, M.G.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noonan, J.J.; Jarzabek, M.; Lincoln, F.A.; Cavanagh, B.L.; Pariag, A.R.; Juric, V.; Young, L.S.; Ligon, K.L.; Jahns, H.; Zheleva, D.; et al. Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma. Cancers 2019, 11, 2005. https://doi.org/10.3390/cancers11122005
Noonan JJ, Jarzabek M, Lincoln FA, Cavanagh BL, Pariag AR, Juric V, Young LS, Ligon KL, Jahns H, Zheleva D, et al. Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma. Cancers. 2019; 11(12):2005. https://doi.org/10.3390/cancers11122005
Chicago/Turabian StyleNoonan, Janis J., Monika Jarzabek, Frank A. Lincoln, Brenton L. Cavanagh, Arhona R. Pariag, Viktorija Juric, Leonie S. Young, Keith L. Ligon, Hanne Jahns, Daniella Zheleva, and et al. 2019. "Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma" Cancers 11, no. 12: 2005. https://doi.org/10.3390/cancers11122005
APA StyleNoonan, J. J., Jarzabek, M., Lincoln, F. A., Cavanagh, B. L., Pariag, A. R., Juric, V., Young, L. S., Ligon, K. L., Jahns, H., Zheleva, D., Prehn, J. H. M., Rehm, M., Byrne, A. T., & Murphy, B. M. (2019). Implementing Patient-Derived Xenografts to Assess the Effectiveness of Cyclin-Dependent Kinase Inhibitors in Glioblastoma. Cancers, 11(12), 2005. https://doi.org/10.3390/cancers11122005