Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL

 ,
, 
Abstract
:1. Introduction
2. Results
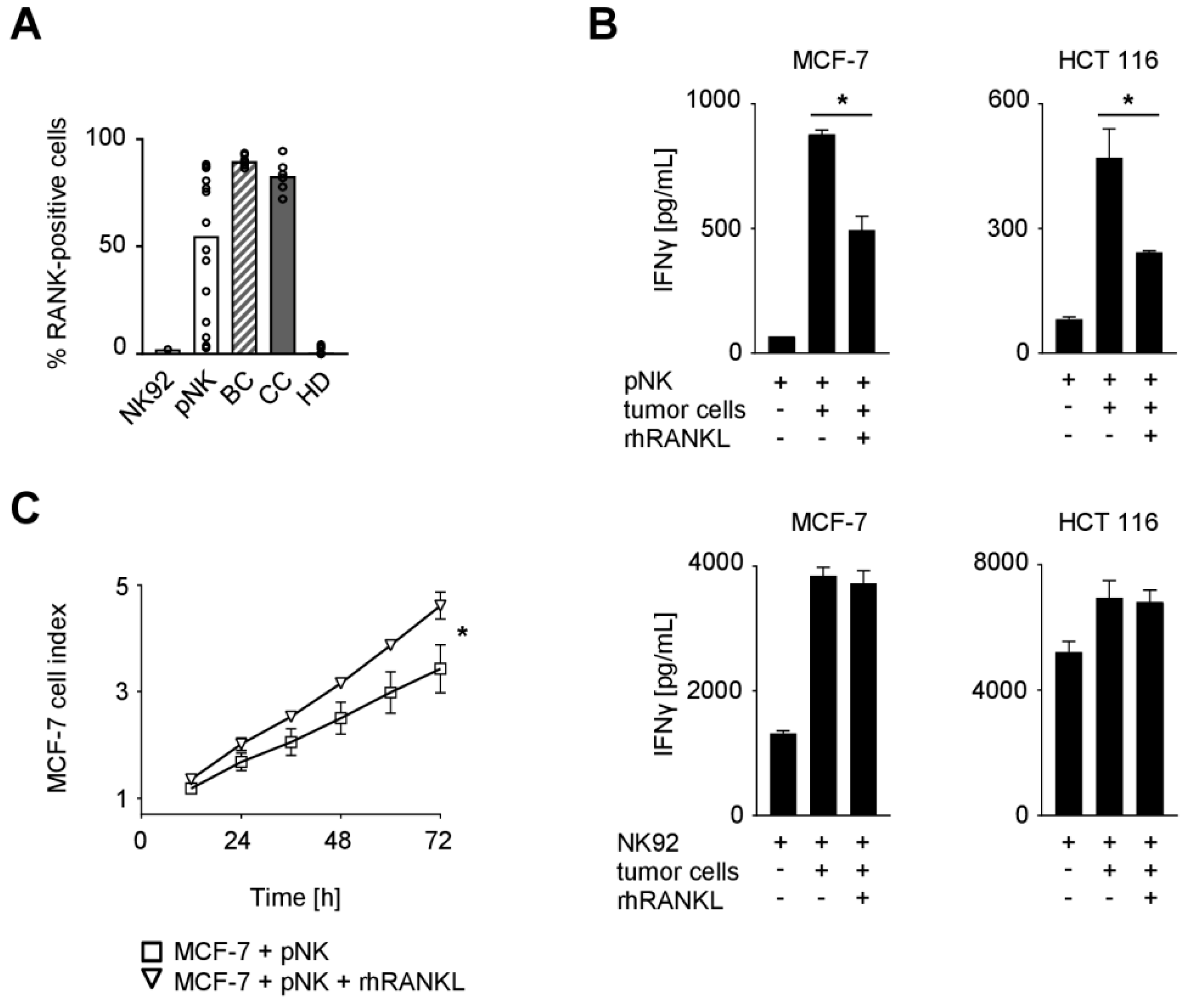
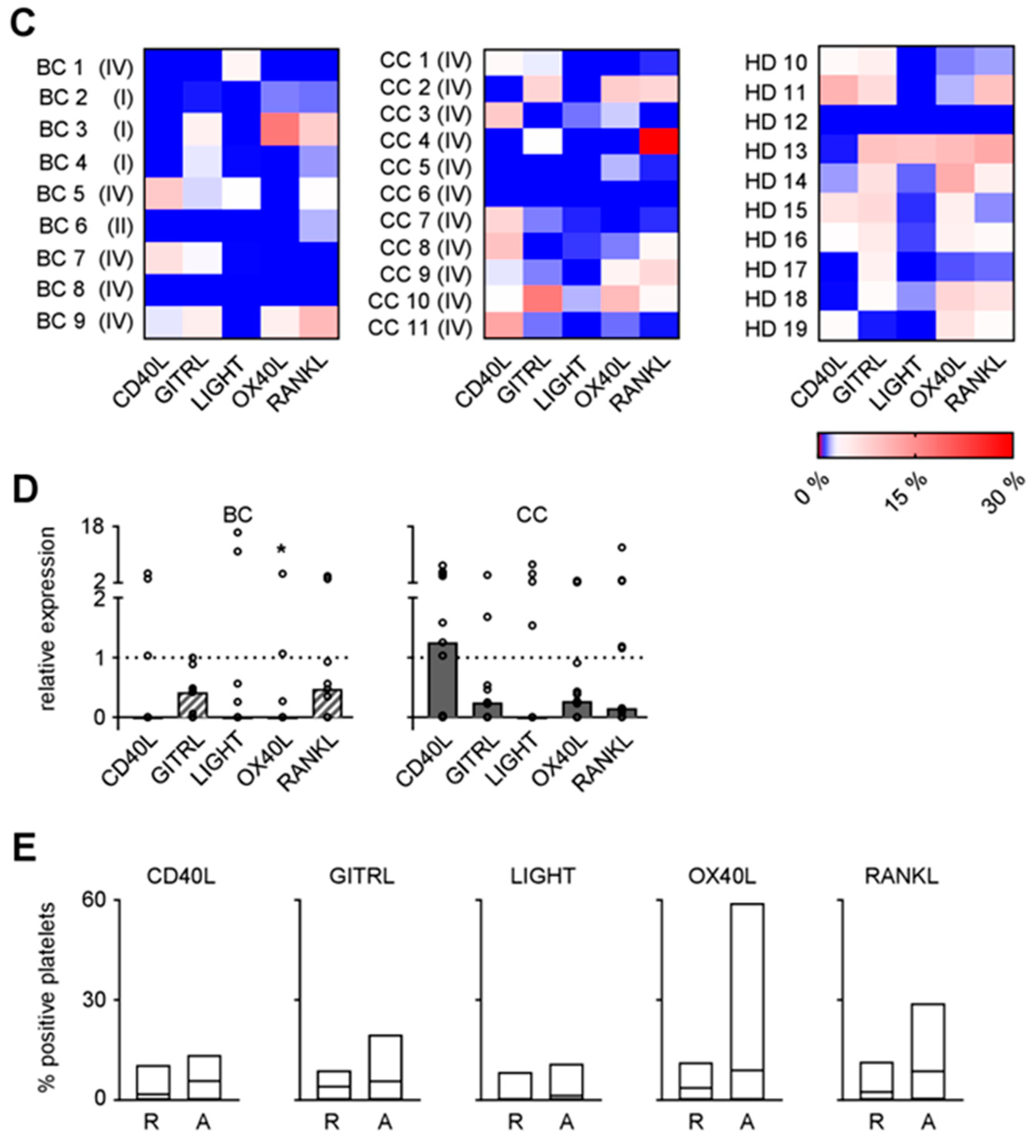
2.1. Expression of TNFR Family Molecules on PBMC Subpopulations
2.2. Functional Effects of the RANK/RANKL Axis in NK Cell Reactivity Against Solid Tumors
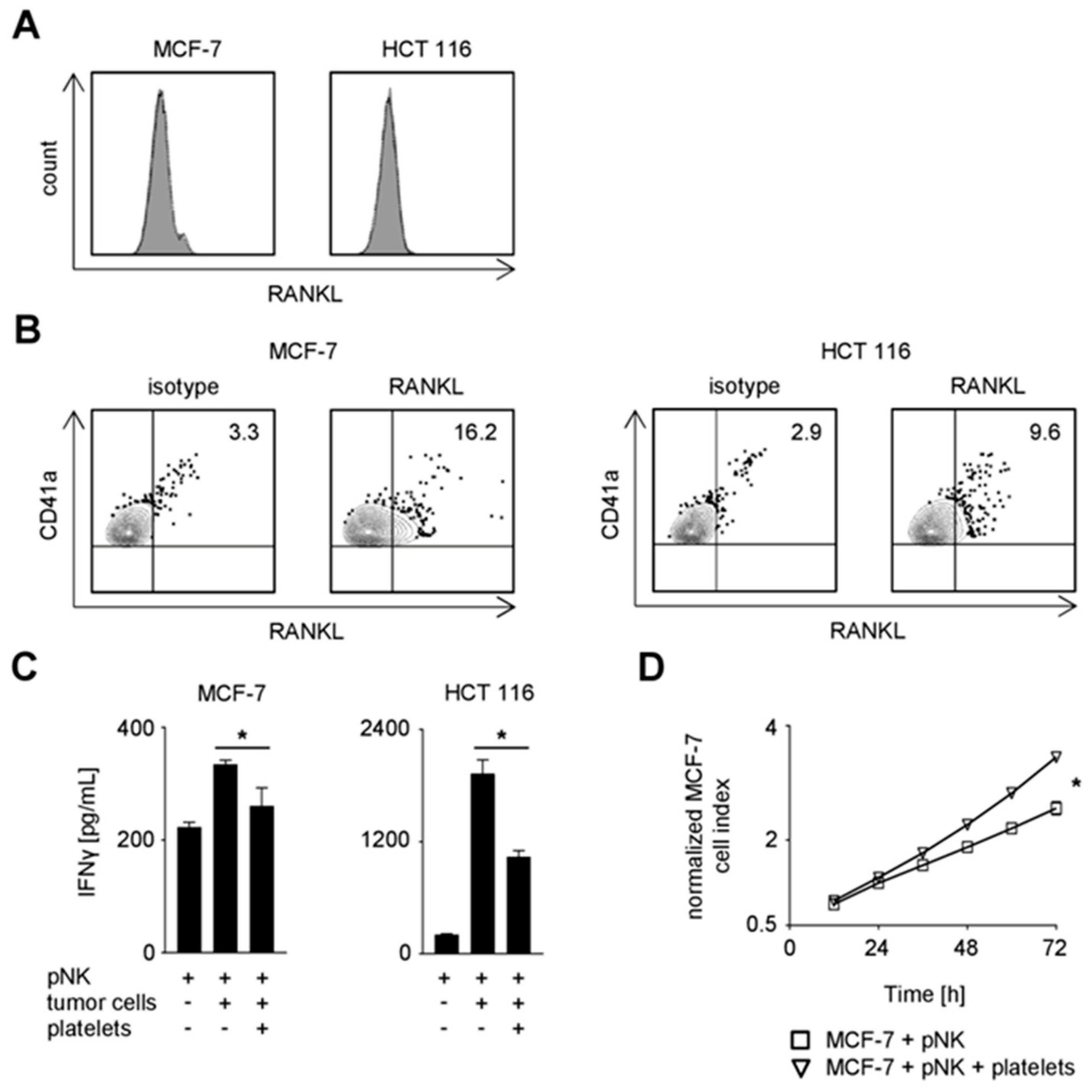
2.3. Expression of TNF Family Members on Platelets
2.4. Functional Effect of Platelet-Derived RANKL on NK Reactivity
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Patients
4.4. Preparation of PBMC, NK Cells, and Platelets
4.5. Treatment of Tumor Cells with Platelets
4.6. Flow Cytometry
4.7. Cytotoxicity Assay
4.8. Determination of IFNγ
4.9. Statistics
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Kopp, H.G.; Salih, H.R. Modulation of Natural Killer Cell Anti-Tumor Reactivity by Platelets. J. Innate Immun. 2011, 4, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Wild, J.; Schmiedel, B.J.; Maurer, A.; Raab, S.; Prokop, L.; Stevanovic, S.; Dorfel, D.; Schneider, P.; Salih, H.R. Neutralization of (NK-cell-derived) B-cell activating factor by Belimumab restores sensitivity of chronic lymphoid leukemia cells to direct and Rituximab-induced NK lysis. Leukemia 2015, 29, 1676–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuebling, T.; Schumacher, C.E.; Hofmann, M.; Hagelstein, I.; Schmiedel, B.J.; Maurer, S.; Federmann, B.; Rothfelder, K.; Roerden, M.; Dorfel, D.; et al. The Immune Checkpoint Modulator OX40 and Its Ligand OX40L in NK-Cell Immunosurveillance and Acute Myeloid Leukemia. Cancer Immunol. Res. 2018, 6, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, Z.; Mahesh, S.P.; Pantanelli, S.; Hwang, F.S.; Siu, W.O.; Nussenblatt, R.B. Glucocorticoid-induced tumor necrosis factor receptor negatively regulates activation of human primary natural killer (NK) cells by blocking proliferative signals and increasing NK cell apoptosis. J. Biol. Chem. 2008, 283, 8202–8210. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Marklin, M.; Kopp, H.G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2018, 7, e1364827. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Raulet, D.H. Activation and self-tolerance of natural killer cells. Immunol. Rev. 2006, 214, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Oergel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC Class I confers a pseudo- normal phenotype to cancer cells that subverts the anti-tumor reactivity of natural killer immune cells. Cancer Res. 2011, 72, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Salih, H.R.; Kopp, H.G. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J. Immunol. 2012, 189, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Inaba, M.; Inagaki-Katashiba, N.; Tanaka, A.; Vien, P.T.; Kibata, K.; Ito, T.; Nomura, S. Platelet-derived RANK ligand enhances CCL17 secretion from dendritic cells mediated by thymic stromal lymphopoietin. Platelets 2015, 26, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, B.J.; Grosse-Hovest, L.; Salih, H.R. A “vicious cycle” of NK-cell immune evasion in acute myeloid leukemia mediated by RANKL? Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmiedel, B.J.; Nuebling, T.; Steinbacher, J.; Malinovska, A.; Wende, C.M.; Azuma, M.; Schneider, P.; Grosse-Hovest, L.; Salih, H.R. Receptor Activator for NF-kappaB Ligand in Acute Myeloid Leukemia: Expression, Function, and Modulation of NK Cell Immunosurveillance. J. Immunol. 2013, 190, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Gruss, H.J.; Dower, S.K. Tumor necrosis factor ligand superfamily: Involvement in the pathology of malignant lymphomas. Blood 1995, 85, 3378–3404. [Google Scholar] [PubMed]
- Handgretinger, R.; Lang, P.; Andre, M.C. Exploitation of natural killer cells for the treatment of acute leukemia. Blood 2016, 127, 3341–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingoni, A.; Sornasse, T.; Cocks, B.G.; Tanaka, Y.; Santoni, A.; Lanier, L.L. Cross-talk between activated human NK cells and CD4+ T cells via OX40-OX40 ligand interactions. J. Immunol. 2004, 173, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Egerdie, B.; Hernandez, T.N.; Feldman, R.; Tammela, T.L.; Saad, F.; Heracek, J.; Szwedowski, M.; Ke, C.; Kupic, A.; et al. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N. Engl. J. Med. 2009, 361, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, L.C.; Neubauer, A.; Heufelder, A.E. Receptor activator of nuclear factor-kappaB ligand and osteoprotegerin: Potential implications for the pathogenesis and treatment of malignant bone diseases. Cancer 2001, 92, 460–470. [Google Scholar] [CrossRef]
- Roodman, G.D.; Dougall, W.C. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat. Rev. 2008, 34, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Gnant, M.; Pfeiler, G.; Dubsky, P.C.; Hubalek, M.; Greil, R.; Jakesz, R.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger-Zeinitzer, E.; et al. The impact of adjuvant denosumab on disease-free survival: Results from 3,425 postmenopausal patients of the ABCSG-18 trial. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Baessler, T.; Krusch, M.; Schmiedel, B.J.; Kloss, M.; Baltz, K.M.; Wacker, A.; Schmetzer, H.M.; Salih, H.R. Glucocorticoid-induced tumor necrosis factor receptor-related protein ligand subverts immunosurveillance of acute myeloid leukemia in humans. Cancer Res. 2009, 69, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Baessler, T.; Charton, J.E.; Schmiedel, B.J.; Grunebach, F.; Krusch, M.; Wacker, A.; Rammensee, H.G.; Salih, H.R. CD137 ligand mediates opposite effects in human and mouse NK cells and impairs NK-cell reactivity against human acute myeloid leukemia cells. Blood 2010, 115, 3058–3069. [Google Scholar] [CrossRef] [PubMed]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Menezes, J.; Knafo, L.; Ahmad, A. Activated human platelets express Fas-L and induce apoptosis in Fas-positive tumor cells. J. Leukoc. Biol. 2001, 69, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Crist, S.A.; Elzey, B.D.; Ludwig, A.T.; Griffith, T.S.; Staack, J.B.; Lentz, S.R.; Ratliff, T.L. Expression of TNF-related apoptosis-inducing ligand (TRAIL) in megakaryocytes and platelets. Exp. Hematol. 2004, 32, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Plantureux, L.; Mege, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Impacts of Cancer on Platelet Production, Activation and Education and Mechanisms of Cancer-Associated Thrombosis. Cancers 2018, 10, 441. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, D.; Benslimane, N.; Al-Zoobi, L.; Hassan, G.; Nadiri, A.; Mourad, W. CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J. Biol. Chem. 2013, 288, 36083–36093. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tsuda, M.; Takahashi, T.; Totsuka, Y.; Shindoh, M.; Ohba, Y. RANKL expression specifically observed in vivo promotes epithelial mesenchymal transition and tumor progression. Am. J. Pathol. 2011, 178, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Willen, L.; Smulski, C.R. Tools and techniques to study ligand-receptor interactions and receptor activation by TNF superfamily members. Methods Enzymol. 2014, 545, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Dennehy, K.M.; Klimosch, S.N.; Steinle, A. Cutting edge: NKp80 uses an atypical hemi-ITAM to trigger NK cytotoxicity. J. Immunol. 2011, 186, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Tam, Y.K.; Maki, G.; Miyagawa, B.; Hennemann, B.; Tonn, T.; Klingemann, H.G. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum. Gene Ther. 1999, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Koerner, S.P.; Andre, M.C.; Leibold, J.S.; Kousis, P.C.; Kubler, A.; Pal, M.; Haen, S.P.; Buhring, H.J.; Grosse-Hovest, L.; Jung, G.; et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia 2016. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BC Patients | CC Patients | |||||
|---|---|---|---|---|---|---|
| Patient Characteristics | Count | % | Patient Characteristics | Count | % | |
| total no. of patients | 9 | 100 | total no. of patients | 11 | 100 | |
| age (years) | age (years) | |||||
| mean | 66 | mean | 63 | |||
| range | 58–81 | range | 47–79 | |||
| sex of patients | sex of patients | |||||
| female | 9 | 100 | female | 4 | 36.4 | |
| male | 0 | 0 | male | 7 | 63.6 | |
| UICC stage | UICC stage | |||||
| I | 3 | 33.3 | I | 0 | 0 | |
| II | 1 | 11.1 | II | 0 | 0 | |
| III | 0 | 0 | III | 0 | 0 | |
| IV | 5 | 55.6 | IV | 11 | 100 | |
| receptor status | cytogenetic | |||||
| Her2 | 5 | 55.6 | Ras mutation | 4 | 36.4 | |
| HR | 6 | 66.7 | MSI | 0 | 0 | |
| Her2 | 1 | 9 | ||||
| PIK3C | 1 | 9 | ||||
| SMAD4 | 1 | 9 | ||||
| TP53 | 2 | 18 | ||||
| BRAF | 1 | 9 | ||||
| no. treatment regimens exposed to | no. treatment regimens exposed to | |||||
| 1 | 5 | 55.6 | 1 | 2 | 18.2 | |
| 2 | 1 | 11.1 | 2 | 5 | 45.4 | |
| ≥3 | 3 | 33.3 | ≥3 | 4 | 36.4 | |
| thromboembolic event | 1 | 11.1 | thromboembolic event | 2 | 18.2 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clar, K.L.; Hinterleitner, C.; Schneider, P.; Salih, H.R.; Maurer, S. Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL. Cancers 2019, 11, 277. https://doi.org/10.3390/cancers11030277
Clar KL, Hinterleitner C, Schneider P, Salih HR, Maurer S. Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL. Cancers. 2019; 11(3):277. https://doi.org/10.3390/cancers11030277
Chicago/Turabian StyleClar, Kim L., Clemens Hinterleitner, Pascal Schneider, Helmut R. Salih, and Stefanie Maurer. 2019. "Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL" Cancers 11, no. 3: 277. https://doi.org/10.3390/cancers11030277
APA StyleClar, K. L., Hinterleitner, C., Schneider, P., Salih, H. R., & Maurer, S. (2019). Inhibition of NK Reactivity Against Solid Tumors by Platelet-Derived RANKL. Cancers, 11(3), 277. https://doi.org/10.3390/cancers11030277