Detecting and Tracking Circulating Tumour DNA Copy Number Profiles during First Line Chemotherapy in Oesophagogastric Adenocarcinoma

 , ,
, ,
Abstract
:1. Introduction
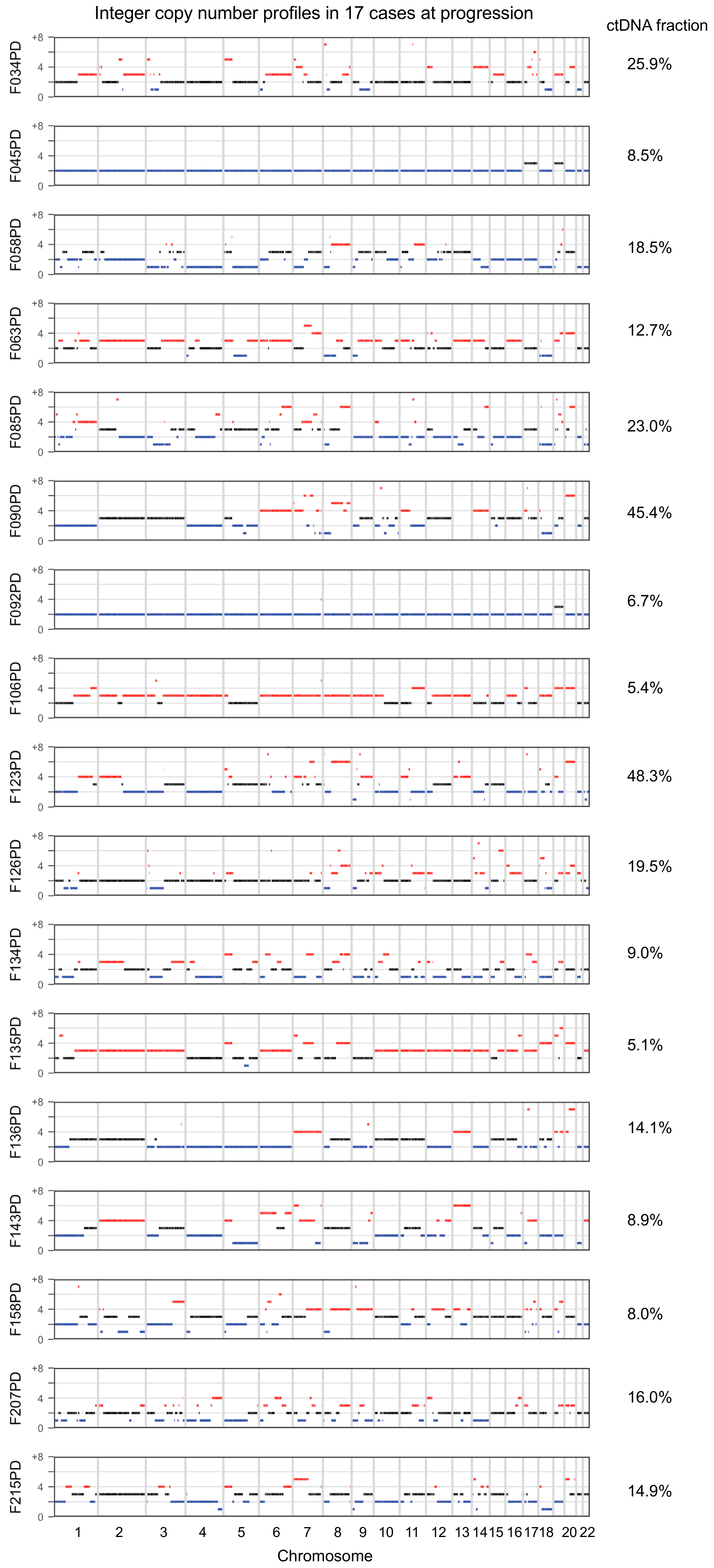
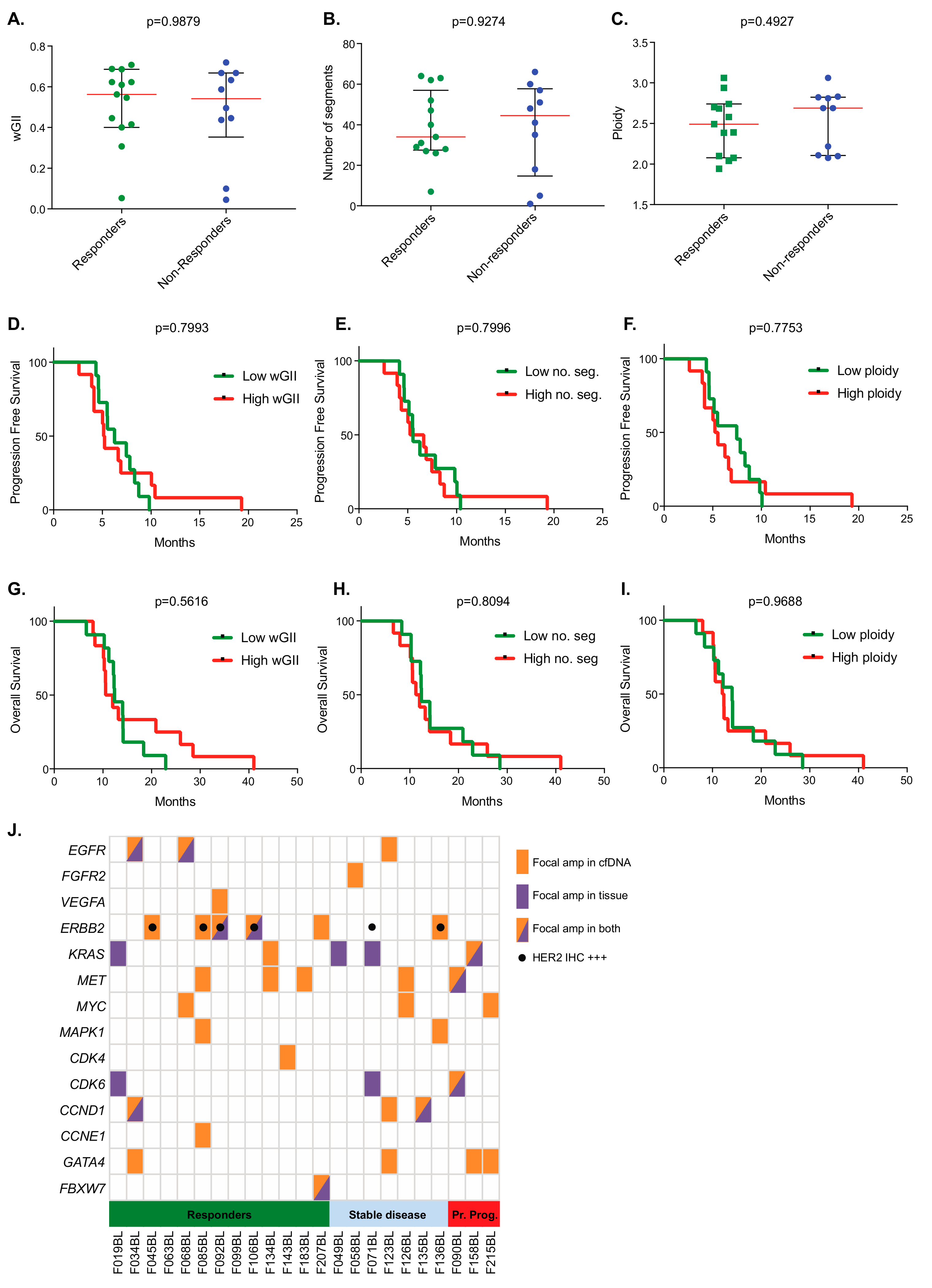
2. Results
3. Discussion
4. Methods
4.1. Trial Design and Sample Collection
4.2. Circulating Free (cf)DNA Extraction and Quantification
4.3. Low-Coverage Whole Genome Sequencing (lcWGS)
4.4. Somatic Copy Number Aberration (SCNA) Analysis
4.5. Survival Analyses by Pre-Treatment Circulating DNA Metrics
4.6. Data Availability
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSMD1 |
| LOC100287015 |
| MCPH1 |
| ANGPT2 |
| CLDN23 |
| MFHAS1 |
| ERI1 |
| MIR4660 |
| PPP1R3B |
| LOC157273 |
| TNKS |
| MIR597 |
| LINC00599 |
| MIR124-1 |
| MSRA |
| PRSS55 |
| RP1L1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.D.; Unverzagt, S.; Grothe, W.; Kleber, G.; Grothey, A.; Haerting, J.; Fleig, W.E. Chemotherapy for advanced gastric cancer. Cochrane database Syst. Rev. 2010, 3, CD004064. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [Green Version]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [Green Version]
- Liang, L.; Fang, J.-Y.; Xu, J. Gastric cancer and gene copy number variation: Emerging cancer drivers for targeted therapy. Oncogene 2016, 35, 1475–1482. [Google Scholar] [CrossRef]
- Labots, M.; Buffart, T.E.; Haan, J.C.; van Grieken, N.C.; Tijssen, M.; van de Velde, C.J.; Grabsch, H.I.; Ylstra, B.; Carvalho, B.; Fijneman, R.J.; et al. High-level copy number gains of established and potential drug target genes in gastric cancer as a lead for treatment development and selection. Cell. Oncol. 2014, 37, 41–52. [Google Scholar] [CrossRef]
- Zhang, Y. Epidemiology of esophageal cancer. World J. Gastroenterol. 2013, 19, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ho, S.S.; Zhang, X.; Pattni, R.; Haraksingh, R.R.; Urban, A.E. Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J. Med. Genet. 2018, 55, 735–743. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Hoffmann, E.M.; Pichler, M.; Gasch, C.; Ulz, P.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Mohan, S.; et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int. J. Cancer 2013, 133, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Alsina, M.; Gullo, I.; Carneiro, F. Intratumoral heterogeneity in gastric cancer: A new challenge to face. Ann. Oncol. 2017, 28, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015, 5, 821–831. [Google Scholar] [CrossRef]
- Lee, H.E.; Park, K.U.; Yoo, S.B.; Nam, S.K.; Park, D.J.; Kim, H.H.; Lee, H.S. Clinical significance of intratumoral HER2 heterogeneity in gastric cancer. Eur. J. Cancer 2013, 49, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.; Smyth, E.; Babina, I.S.; Herrera-Abreu, M.T.; Tarazona, N.; Peckitt, C.; Kilgour, E.; Smith, N.R.; Geh, C.; Rooney, C.; et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 2016, 6, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Petty, R.D.; Dahle-Smith, A.; Stevenson, D.A.J.; Osborne, A.; Massie, D.; Clark, C.; Murray, G.I.; Dutton, S.J.; Roberts, C.; Chong, I.Y.; et al. Gefitinib and EGFR Gene Copy Number Aberrations in Esophageal Cancer. J. Clin. Oncol. 2017, 35, 2279–2287. [Google Scholar] [CrossRef]
- Gao, J.; Wang, H.; Zang, W.; Li, B.; Rao, G.; Li, L.; Yu, Y.; Li, Z.; Dong, B.; Lu, Z.; et al. Circulating tumor DNA functions as an alternative for tissue to overcome tumor heterogeneity in advanced gastric cancer. Cancer Sci. 2017, 108, 1881–1887. [Google Scholar] [CrossRef]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef]
- Chaplet, M.; Rai, R.; Jackson-Bernitsas, D.; Li, K.; Lin, S.Y. BRIT1/MCPH1: A guardian of genome and an enemy of tumors. Cell Cycle 2006, 5, 2579–2583. [Google Scholar] [CrossRef]
- Wei, Q.; Wang, X.; An, X.; Han, Q.; Meng, L.; Cao, W.L.Z. Effects of MCPH1 silencing on proliferation, apoptosis, and chemo-sensitivity of non-small cell lung cancer cells. Int. J. Clin. Exp. Med. 2018, 11, 6583–6595. [Google Scholar]
- Dulak, A.M.; Schumacher, S.E.; Van Lieshout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.H.; Saksena, G.; Baca, S.C.; Baselga, J.; et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014, 4, 175–185. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Villegas, E.V.; Rondón-Lagos, M. The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2018, 10, 4. [Google Scholar] [CrossRef]
- Burrell, R.A.; Mcgranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Endesfelder, D.; Burrell, R.A.; Kanu, N.; McGranahan, N.; Howell, M.; Parker, P.J.; Downward, J.; Swanton, C.; Kschischo, M. Chromosomal instability selects gene copy-number variants encoding core regulators of proliferation in ER+ Breast cancer. Cancer Res. 2014, 74, 4853–4863. [Google Scholar] [CrossRef]
- Baslan, T.; Kendall, J.; Rodgers, L.; Cox, H.; Riggs, M.; Stepansky, A.; Troge, J.; Ravi, K.; Esposito, D.; Lakshmi, B.; et al. Genome-wide copy number analysis of single cells. Nat. Protoc. 2012, 7, 1024–1041. [Google Scholar] [CrossRef]
- Moorcraft, S.Y.; Gonzalez de Castro, D.; Cunningham, D.; Jones, T.; Walker, B.A.; Peckitt, C.; Yuan, L.C.; Frampton, M.; Begum, R.; Eltahir, Z.; et al. Investigating the feasibility of tumour molecular profiling in gastrointestinal malignancies in routine clinical practice. Ann. Oncol. 2018, 29, 230–236. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Sanchez-Vega, F.; Jonsson, P.; Chatila, W.K.; Hechtman, J.F.; Ku, G.Y.; Riches, J.C.; Tuvy, Y.; Kundra, R.; Bouvier, N.; et al. Genetic Predictors of Response to Systemic Therapy in Esophagogastric Cancer. Cancer Discov. 2018, 8, 49–58. [Google Scholar] [CrossRef]
- Fang, W.-L.; Lan, Y.-T.; Huang, K.-H.; Liu, C.-A.; Hung, Y.-P.; Lin, C.-H.; Jhang, F.-Y.; Chang, S.-C.; Chen, M.-H.; Chao, Y.; et al. Clinical significance of circulating plasma DNA in gastric cancer. Int. J. Cancer 2016, 138, 2974–2983. [Google Scholar] [CrossRef] [Green Version]
- Saluja, H.; Karapetis, C.S.; Pedersen, S.K.; Young, G.P.; Symonds, E.L. The Use of Circulating Tumor DNA for Prognosis of Gastrointestinal Cancers. Front. Oncol. 2018, 8, 275. [Google Scholar] [CrossRef]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Fuca, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of primary resistance to trastuzumab in HER2-positive metastatic gastric cancer patients: The AMNESIA case-control study. Clin. Cancer Res. 2018, 24, 1082–1089. [Google Scholar] [CrossRef]
- Menyhart, O.; Santarpia, L.; Gyorffy, B. A Comprehensive Outline of Trastuzumab Resistance Biomarkers in HER2 Overexpressing Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 665–683. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Welz, H.; Marienfeld, R.; Schmidt, S.A.; Kleger, A.; Ettrich, T.J.; Seufferlein, T. Treatment monitoring in metastatic colorectal cancer patients by quantification and KRAS genotyping of circulating cell-free DNA. PLoS ONE 2017, 12, e0174308. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-S.; Liu, Z.-X.; Lu, Y.-X.; Bao, H.; Wu, X.; Zeng, Z.-L.; Liu, Z.; Zhao, Q.; He, C.-Y.; Lu, J.-H.; et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut 2018. [Google Scholar] [CrossRef]
- Zheng, X.; Fan, L.; Zhou, P.; Ma, H.; Huang, S.; Yu, D.; Zhao, L.; Yang, S.; Liu, J.; Huang, A.; et al. Detection of Circulating Tumor Cells and Circulating Tumor Microemboli in Gastric Cancer. Transl. Oncol. 2017, 10, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Matsusaka, S.; Chin, K.; Mikuniya, M.; Minowa, S.; Takayama, T.; Shibata, H.; Kuniyoshi, R.; Ogura, M.; Terui, Y.; et al. Detection of HER2 Amplification in Circulating Tumor Cells of HER2-Negative Gastric Cancer Patients. Target. Oncol. 2017, 12, 341–351. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Antonarakis, E.S.; Gjyrezi, A.; Galletti, G.; Kim, S.; Worroll, D.; Stewart, J.; Zaher, A.; Szatrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and ARV 567Es in circulating tumor cells correlates with outcomes to taxane therapy in men with metastatic prostate cancer treated in taxynergy. Clin. Cancer Res. 2019, 25, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Lallo, A.; Frese, K.K.; Morrow, C.J.; Sloane, R.; Gulati, S.; Schenk, M.W.; Trapani, F.; Simms, N.; Galvin, M.; Brown, S.; et al. The combination of the PARP inhibitor olaparib and the WEE1 Inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin. Cancer Res. 2018, 24, 5153–5164. [Google Scholar] [CrossRef] [PubMed]
- Lallo, A.; Gulati, S.; Schenk, M.W.; Khandelwal, G.; Berglund, U.W.; Pateras, I.S.; Chester, C.P.E.; Pham, T.M.; Kalderen, C.; Frese, K.K.; et al. Ex vivo culture of cells derived from circulating tumour cell xenograft to support small cell lung cancer research and experimental therapeutics. Br. J. Pharmacol. 2019, 176, 436–450. [Google Scholar] [CrossRef]
- Fu, M.; Gu, J.; Jiang, P.; Qian, H.; Xu, W.; Zhang, X. Exosomes in gastric cancer: Roles, mechanisms, and applications. Mol. Cancer 2019, 18, 41. [Google Scholar] [CrossRef]
- Mansukhani, S.; Barber, L.J.; Kleftogiannis, D.; Moorcraft, S.Y.; Davidson, M.; Woolston, A.; Proszek, P.Z.; Griffiths, B.; Fenwick, K.; Herman, B.; et al. Ultra-Sensitive mutation detection and genome-wide DNA copy number reconstruction by error- corrected circulating tumor DNA sequencing. Clin. Chem. 2018, 64, 1626–1635. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.; Roth, A.; Lai, D.; Bashashati, A.; Ding, J.; Goya, R.; Giuliany, R.; Rosner, J.; Oloumi, A.; Shumansky, K.; et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012, 22, 1995–2007. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, G.; Liestøl, K.; Van Loo, P.; Moen Vollan, H.K.; Eide, M.B.; Rueda, O.M.; Chin, S.F.; Russell, R.; Baumbusch, L.O.; Caldas, C.; et al. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC Genom. 2012, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24. [Google Scholar] [CrossRef]



| Histopathological Variable | ||
|---|---|---|
| Number of Cases: | 30 | |
| Anatomic site of primary: | Gastric | 6 (20%) |
| OGJ/oesophageal | 24 (80%) | |
| Histological subtype: | Intestinal | 28 (93%) |
| Diffuse | 2 (7%) | |
| Clinical stage at presentation: | Locally advanced | 3 (10%) |
| Metastatic | 27 (90%) | |
| HER2 status *: | Positive | 6 (20%) |
| Negative | 24 (80%) | |
| First line chemotherapy: | Platinum/fluoropyrimidine doublet | 9 (30%) |
| Doublet+ anthracycline | 15 (50%) | |
| Doublet+ trastuzumab | 6 (20%) | |
| Metastatic sites: Liver | Yes | 16 (53%) |
| No | 14 (47%) | |
| Peritoneal | Yes | 6 (20%) |
| No | 24 (80%) | |
| Lung | Yes | 8 (27%) |
| No | 22 (73%) | |
| Number of metastatic organ sites: | 0–1 | 22 (73%) |
| ≥2 | 8 (27%) | |
| Primary tumour in situ: | Yes | 23 (77%) |
| No | 7 (23%) | |
| CA19-9 secretor: | Yes | 15 (50%) |
| No | 15 (50%) | |
| Histopathological Variable | n | Median cfDNA Concentration (ng/mL Plasma) | p-Value | Median ctDNA Fraction (%) | p-Value | Median ctDNA Concentration (ng/mL Plasma) | p-Value | |
|---|---|---|---|---|---|---|---|---|
| Primary tumour in situ | Yes | 23 | 9.66 | 0.0027 | 9.10 | 0.0046 | 2.14 | <0.0001 |
| No | 7 | 4.81 | 0.00 | 0.00 | ||||
| Liver metastases present | Yes | 16 | 10.09 | 0.1306 | 18.01 | 0.0043 | 2.18 | 0.0099 |
| No | 14 | 6.80 | 7.23 | 0.35 | ||||
| Primary tumour anatomic site | Gastric | 6 | 8.65 | 0.8996 | 3.33 | 0.0103 | 0.24 | 0.1401 |
| Nongastric | 24 | 9.05 | 9.31 | 0.84 | ||||
| No. of metastatic organ sites | 0–1 | 22 | 8.31 | 0.5042 | 7.77 | 0.1528 | 0.47 | 0.9814 |
| ≥2 | 8 | 1.22 | 14.47 | 0.58 | ||||
| HER2 status | Positive | 6 | 11.22 | 0.3739 | 8.81 | 0.4595 | 2.25 | 0.1713 |
| Negative | 24 | 8.32 | 8.22 | 0.47 | ||||
| CA19-9 secretion | Yes | 15 | 9.21 | 0.9999 | 8.10 | 0.5640 | 0.61 | 0.7733 |
| No | 15 | 8.54 | 9.02 | 0.78 | ||||
| Paired Pretreatment and Progression Cases | n | Median ctDNA Fraction (%) | p-Value | |
|---|---|---|---|---|
| All paired cases | Pretreatment | 20 | 15.18 | 0.1567 |
| Progression | 20 | 8.72 | ||
| Initial radiological response followed by progression to chemotherapy: ‘primary responders’ | Pretreatment | 12 | 17.00 | 0.0200 |
| Progression | 12 | 7.59 | ||
| Stable disease or primary radiological progression to chemotherapy: ‘primary nonresponders’ | Pretreatment | 8 | 11.27 | 0.7984 |
| Progression | 8 | 13.58 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson, M.; Barber, L.J.; Woolston, A.; Cafferkey, C.; Mansukhani, S.; Griffiths, B.; Moorcraft, S.-Y.; Rana, I.; Begum, R.; Assiotis, I.; et al. Detecting and Tracking Circulating Tumour DNA Copy Number Profiles during First Line Chemotherapy in Oesophagogastric Adenocarcinoma. Cancers 2019, 11, 736. https://doi.org/10.3390/cancers11050736
Davidson M, Barber LJ, Woolston A, Cafferkey C, Mansukhani S, Griffiths B, Moorcraft S-Y, Rana I, Begum R, Assiotis I, et al. Detecting and Tracking Circulating Tumour DNA Copy Number Profiles during First Line Chemotherapy in Oesophagogastric Adenocarcinoma. Cancers. 2019; 11(5):736. https://doi.org/10.3390/cancers11050736
Chicago/Turabian StyleDavidson, Michael, Louise J. Barber, Andrew Woolston, Catherine Cafferkey, Sonia Mansukhani, Beatrice Griffiths, Sing-Yu Moorcraft, Isma Rana, Ruwaida Begum, Ioannis Assiotis, and et al. 2019. "Detecting and Tracking Circulating Tumour DNA Copy Number Profiles during First Line Chemotherapy in Oesophagogastric Adenocarcinoma" Cancers 11, no. 5: 736. https://doi.org/10.3390/cancers11050736