Molecular Profiling of Atypical Tenosynovial Giant Cell Tumors Reveals Novel Non-CSF1 Fusions


Abstract
:1. Introduction
2. Results
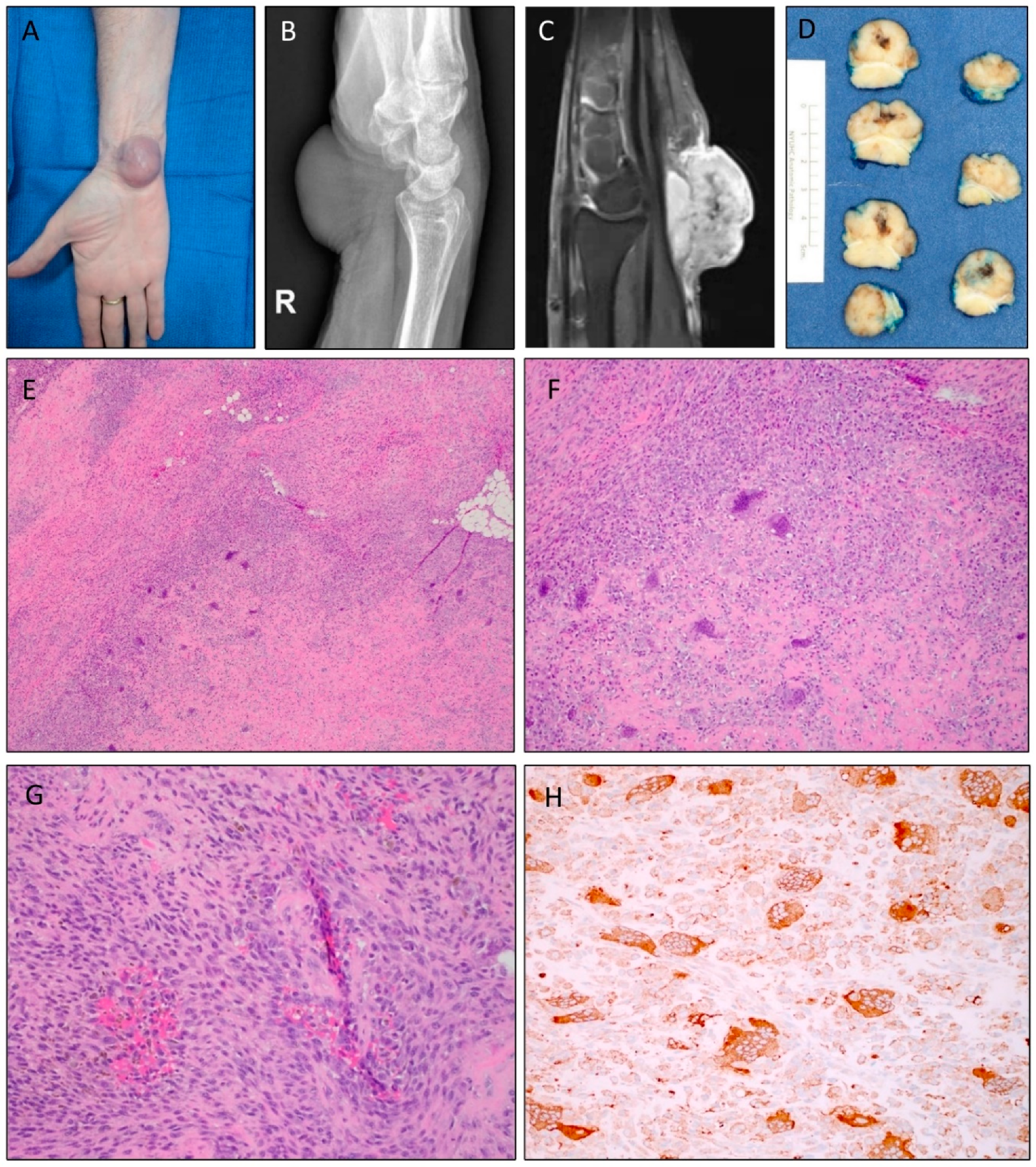
2.1. Clinical Presentation and Morphology of Atypical TGCTs
2.2. Identification of Novel Gene Fusions in Atypical TGCTs
2.3. Detection of New CSF1 Fusion Partners in Conventional TGCTs
3. Discussion
4. Materials and Methods
4.1. Case Selection
4.2. Immunohistochemistry
4.3. Detection and Validation of Fusions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Somerhausen, N.S.; Fletcher, C.D. Diffuse-type giant cell tumor: Clinicopathologic and immunohistochemical analysis of 50 cases with extraarticular disease. Am. J. Surg. Pathol. 2000, 24, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, M.; Hashimoto, H.; Tsuneyoshi, M.; Enjoji, M. Giant cell tumor of the tendon sheath (nodular tenosynovitis): A study of 207 cases to compare the large joint group with the common digit group. Cancer 1986, 57, 875–884. [Google Scholar] [CrossRef]
- Monaghan, H.; Salter, D.M.; Al-Nafussi, A. Giant cell tumour of tendon sheath (localised nodular tenosynovitis): Clinicopathological features of 71 cases. J. Clin. Pathol. 2001, 54, 404–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotwal, P.P.; Gupta, V.; Malhotra, R. Giant-cell tumour of the tendon sheath. Is radiotherapy indicated to prevent recurrence after surgery? J. Bone Jt. Surg. Br. 2000, 82, 571–573. [Google Scholar] [CrossRef]
- Nilsson, M.; Hoglund, M.; Panagopoulos, I.; Sciot, R.; Dal Cin, P.; Debiec-Rychter, M.; Mertens, F.; Mandahl, N. Molecular cytogenetic mapping of recurrent chromosomal breakpoints in tenosynovial giant cell tumors. Virchows Arch. 2002, 441, 475–480. [Google Scholar] [PubMed]
- West, R.B.; Rubin, B.P.; Miller, M.A.; Subramanian, S.; Kaygusuz, G.; Montgomery, K.; Zhu, S.; Marinelli, R.J.; De Luca, A.; Downs-Kelly, E.; et al. A landscape effect in tenosynovial giant-cell tumor from activation of csf1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Cupp, J.S.; Miller, M.A.; Montgomery, K.D.; Nielsen, T.O.; O’Connell, J.X.; Huntsman, D.; van de Rijn, M.; Gilks, C.B.; West, R.B. Translocation and expression of csf1 in pigmented villonodular synovitis, tenosynovial giant cell tumor, rheumatoid arthritis and other reactive synovitides. Am. J. Surg. Pathol. 2007, 31, 970–976. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Brandal, P.; Gorunova, L.; Bjerkehagen, B.; Heim, S. Novel csf1-s100a10 fusion gene and csf1 transcript identified by RNA sequencing in tenosynovial giant cell tumors. Int. J. Oncol. 2014, 44, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Sekine, S.; Kiyono, T.; Ryo, E.; Ogawa, R.; Wakai, S.; Ichikawa, H.; Suzuki, K.; Arai, S.; Tsuta, K.; Ishida, M.; et al. Recurrent yap1-maml2 and yap1-nutm1 fusions in poroma and porocarcinoma. J. Clin. Investig. 2019, 130, 3827–3832. [Google Scholar] [CrossRef]
- Tsuda, Y.; Hirata, M.; Katayama, K.; Motoi, T.; Matsubara, D.; Oda, Y.; Fujita, M.; Kobayashi, H.; Kawano, H.; Nishida, Y.; et al. Massively parallel sequencing of tenosynovial giant cell tumors reveals novel csf1 fusion transcripts and novel somatic cbl mutations. Int. J. Cancer 2019, 145, 3276–3284. [Google Scholar] [CrossRef]
- Al-Ibraheemi, A.; Ahrens, W.A.; Fritchie, K.; Dong, J.; Oliveira, A.M.; Balzer, B.; Folpe, A.L. Malignant tenosynovial giant cell tumor: The true “synovial sarcoma?” A clinicopathologic, immunohistochemical, and molecular cytogenetic study of 10 cases, supporting origin from synoviocytes. Mod. Pathol. 2019, 32, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Goldblum, J.R.; Folpe, A.L.; Weiss, S.W. Enzinger and Weiss’s Soft Tissue Tumors, 6th ed.; Elsevier: Philadelphia, PA, USA, 2014. [Google Scholar]
- Jo, V.Y.; Fletcher, C.D. Who classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L. Phosphaturic mesenchymal tumors: A review and update. Semin. Diagn. Pathol. 2019, 36, 260–268. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.X.; Wehrli, B.M.; Nielsen, G.P.; Rosenberg, A.E. Giant cell tumors of soft tissue: A clinicopathologic study of 18 benign and malignant tumors. Am. J. Surg. Pathol. 2000, 24, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.; Trojanowska, M.; Watson, D.K. Ets proteins in biological control and cancer. J. Cell. Biochem. 2004, 91, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, T.; Yamada, T. Molecular biology of the ETS family of transcription factors. Gene 2003, 303, 11–34. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the tmprss2-erg gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Demichelis, F.; Fall, K.; Perner, S.; Andren, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. Tmprss2: Erg gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef] [Green Version]
- Perner, S.; Demichelis, F.; Beroukhim, R.; Schmidt, F.H.; Mosquera, J.M.; Setlur, S.; Tchinda, J.; Tomlins, S.A.; Hofer, M.D.; Pienta, K.G.; et al. Tmprss2:Erg fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res. 2006, 66, 8337–8341. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.; Kalsaas, A.H.; Buechner, J.; Busund, L.T. Ewing sarcoma-peripheral neuroectodermal tumor of the kidney with a fus-erg fusion transcript. Cancer Genet. Cytogenet. 2009, 194, 53–57. [Google Scholar] [CrossRef]
- Ginsberg, J.P.; de Alava, E.; Ladanyi, M.; Wexler, L.H.; Kovar, H.; Paulussen, M.; Zoubek, A.; Dockhorn-Dworniczak, B.; Juergens, H.; Wunder, J.S.; et al. Ews-fli1 and ews-erg gene fusions are associated with similar clinical phenotypes in ewing’s sarcoma. J. Clin. Oncol. 1999, 17, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Shing, D.C.; McMullan, D.J.; Roberts, P.; Smith, K.; Chin, S.F.; Nicholson, J.; Tillman, R.M.; Ramani, P.; Cullinane, C.; Coleman, N. Fus/erg gene fusions in ewing’s tumors. Cancer Res. 2003, 63, 4568–4576. [Google Scholar] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. Cosmic: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COSMIC. Available online: https://cancer.sanger.ac.uk/cosmic/fusion/overview?Fid=2778&gid=1538 (accessed on 3 October 2019).
- Chen, S.; Deniz, K.; Sung, Y.S.; Zhang, L.; Dry, S.; Antonescu, C.R. Ewing sarcoma with erg gene rearrangements: A molecular study focusing on the prevalence of fus-erg and common pitfalls in detecting ewsr1-erg fusions by fish. Genes Chromosomes Cancer 2016, 55, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Giordano, F.; Kuznetsova, A.; Suja, J.A.; Lander, A.D.; Calof, A.L.; Strom, L. Localisation of the smc loading complex nipbl/mau2 during mammalian meiotic prophase i. Chromosoma 2014, 123, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.H.; Devoto, M.; Jackson, L.G.; et al. Nipbl mutational analysis in 120 individuals with cornelia de lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieslik, M.; Pitchiaya, S.; Chakravarthi, B.; Cao, X.; Jing, X.; et al. Development of peptidomimetic inhibitors of the erg gene fusion product in prostate cancer. Cancer Cell 2017, 31, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Satoh, H.; Yoshida, M.C.; Matsushime, H.; Shibuya, M.; Sasaki, M. Regional localization of the human c-ros-1 on 6q22 and flt on 13q12. Jpn. J. Cancer Res. GANN 1987, 78, 772–775. [Google Scholar]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ou, S.H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. Ros1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Jeng, Y.M.; Su, S.Y.; Wu, C.T.; Tsai, K.S.; Lee, C.H.; Lin, C.Y.; Carter, J.M.; Huang, J.W.; Chen, S.H.; et al. Identification of a novel fn1-fgfr1 genetic fusion as a frequent event in phosphaturic mesenchymal tumour. J. Pathol. 2015, 235, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Yoshida, A.; Hosoda, F.; Nakamura, H.; Hama, N.; Ogura, K.; Yoshida, A.; Fujiwara, T.; Arai, Y.; Toguchida, J.; et al. Unique mutation portraits and frequent col2a1 gene alteration in chondrosarcoma. Genome Res. 2014, 24, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puls, F.; Hofvander, J.; Magnusson, L.; Nilsson, J.; Haywood, E.; Sumathi, V.P.; Mangham, D.C.; Kindblom, L.G.; Mertens, F. Fn1-egf gene fusions are recurrent in calcifying aponeurotic fibroma. J. Pathol. 2016, 238, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, V.; Kohlmann, A.; Klein, H.U.; Schindela, S.; Schnittger, S.; Dicker, F.; Dugas, M.; Kern, W.; Haferlach, T.; Haferlach, C. Targeted next-generation sequencing detects point mutations, insertions, deletions and balanced chromosomal rearrangements as well as identifies novel leukemia-specific fusion genes in a single procedure. Leukemia 2011, 25, 671–680. [Google Scholar] [CrossRef]
- Alexiev, B.A.; Tumer, Y.; Yang, G.Y. Malignant tenosynovial giant cell tumor with CDKN2A/B genomic alteration: A histological, immunohistochemical, and molecular study. Hum. Pathol. 2017, 63, 144–148. [Google Scholar] [CrossRef]
- Møller, M.B.; Ino, Y.; Gerdes, A.M.; Skjødt, K.; Louis, D.N.; Pedersen, N.T. Aberrations of the p53 pathway components p53, MDM2 and CDKN2A appear independent in diffuse large B cell lymphoma. Leukemia 1999, 13, 453–459. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Tumor Type | Fusion | Location/Laterality | Size (cm) | Sex | Age (Years) | Local Recurrence | Duration to Latest Recurrence (Months) | Follow-Up after Latest Excision/Recurrence (Months) | Atypical Features |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Atypical TGCT | NIPBL-ERG | Wrist/Right | 3.2 | Female | 69 | Yes; multiple | 7 | 9 | Prominent cellularity; increased mitotic activity; spindle cells |
| 2 | Atypical TGCT | FN1-ROS1 | Wrist/Left | 2; fragmented | Female | 46 | Yes; multiple | 104 | 32 | Hyperchromatic nuclei; symplastic change; spindle cells |
| 3 | Atypical TGCT | YAP1-MAML2 | Ankle/Left | 3.8 | Female | 64 | No | N/A | 4 | Enlarged nuclei; virocyte-like nucleoli; myxoid change; spindle cells |
| 4 | TGCT | CSF1-COL6A3 | Arm/Left | 1.5 | Female | 14 | No | N/A | 14 | N/A |
| 5 | TGCT | CSF1-KCNMA1 | Knee/Right | 2.5 | Male | 43 | No | N/A | 15 | N/A |
| 6 | TGCT | CSF1-FN1 | Knee/Right | 2.5 | Male | 29 | No | N/A | 15 | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vougiouklakis, T.; Shen, G.; Feng, X.; Hoda, S.T.; Jour, G. Molecular Profiling of Atypical Tenosynovial Giant Cell Tumors Reveals Novel Non-CSF1 Fusions. Cancers 2020, 12, 100. https://doi.org/10.3390/cancers12010100
Vougiouklakis T, Shen G, Feng X, Hoda ST, Jour G. Molecular Profiling of Atypical Tenosynovial Giant Cell Tumors Reveals Novel Non-CSF1 Fusions. Cancers. 2020; 12(1):100. https://doi.org/10.3390/cancers12010100
Chicago/Turabian StyleVougiouklakis, Theodore, Guomiao Shen, Xiaojun Feng, Syed T. Hoda, and George Jour. 2020. "Molecular Profiling of Atypical Tenosynovial Giant Cell Tumors Reveals Novel Non-CSF1 Fusions" Cancers 12, no. 1: 100. https://doi.org/10.3390/cancers12010100