Does Direct and Indirect Exposure to Ionising Radiation Influence the Metastatic Potential of Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Signalling Molecules and Radiation Response
3. Metastasis
3.1. Hypoxia and Angiogenesis
3.2. Disassociation of Cancer Cells from the Primary Tumour Mass and Epithelial-Mesenchymal Transition (EMT)
3.3. Invasion of the Basement Membrane and Extracellular Matrix and Motility
3.4. Intravasation and Hematogenous/Lymphogenous Dissemination
3.5. Extravasation and Invasion of the Basement Membrane and Extracellular Matrix
3.6. Establishment of Tumour at a New Site
4. Potential Involvement of Glycosylation in Metastasis
4.1. Increased Complexity and Branching of N-glycans
4.2. Truncation of O-glycans
4.3. Alterations in Sialylation
5. Changes in Cancer Cell Metastatic Capacity Post-Irradiation
5.1. The Effect of Radiation on Angiogenesis
5.2. The Effect of Radiation on Cancer Cell Motility, Invasion, and EMT
5.3. The Effect of Radiation on Normal and Cancer Cell Glycosylation
5.4. Metastasis Following Radiotherapy in the Clinical Setting
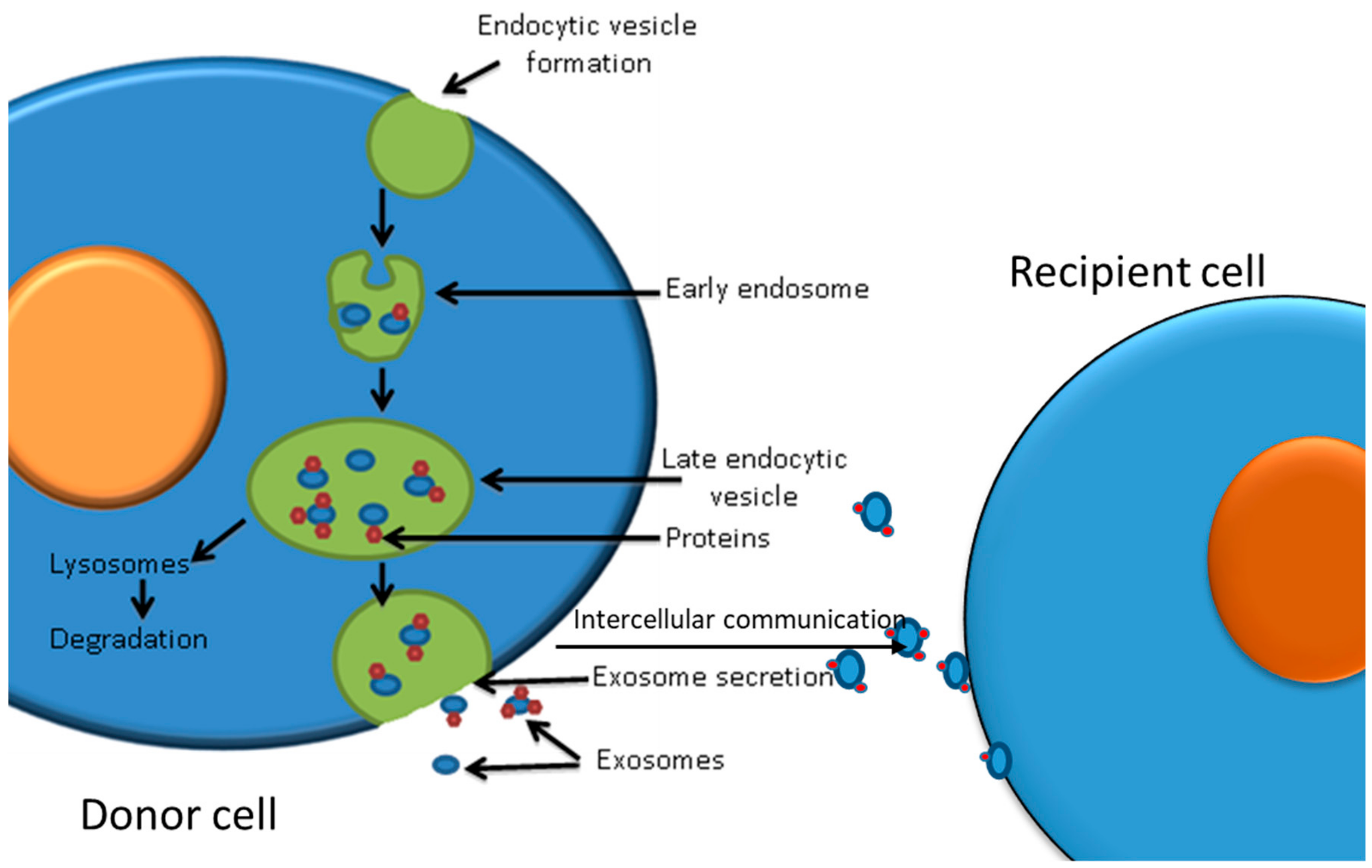
6. Exosomes and the Effect of Post-Irradiation Exosomes on Cancer Cells
6.1. Exosomes as Mediators of Cell–Cell Communication
6.2. Exosomes Involvement in Enhancing Tumour Progression and Metastasis
6.3. The Potential of Exosomes to Enhance the Effectiveness of Radiation Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ronckers, C.M.; Erdmann, C.A.; Land, C.E. Radiation and breast cancer: A review of current evidence. Breast Cancer Res. 2005, 7, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, D.L.; Mattson, A.M.; Holmberg, E.; Shore, R.; Hildreth, N.; Boice, J.D., Jr. Radiation effects on breast cancer risk: A pooled analysis of eight cohorts. Radiat. Res. 2002, 158, 220–235. [Google Scholar] [CrossRef]
- Howard, B.A.; Gusterson, B.A. Human breast development. J. Mammary Gland Biol. Neoplasia 2002, 5, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Russo, I.H. Toward a unified concept of mammary carcinogenesis. Prog. Clin. Biol. Res. 1997, 396, 1–16. [Google Scholar] [PubMed]
- Moolkavgar, S.H.; Day, N.E.; Stevens, R.G. Two-stage model for carcinogenesis: Epidemiology of breast cancer in females. J. Natl. Cancer Inst. 1980, 65, 559–569. [Google Scholar]
- BEIR. The Effects on Populations of Exposure to Low Levels of Ionizing Radiation; Report of the Advisory Committee on the Bio- logical Effects of Ionizing Radiations (BEIR); National Academy of Sciences: Washington, DC, USA, 1980; pp. 136–145. [Google Scholar]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Miki, Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004, 95, 866–871. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Van Assen-Bolt, A.J.; Van Waarde-Verhagen, M.A.; Sijmons, R.H.; Van der Hout, A.H.; Bauch, T.; Streffer, C.; Kampinga, H.H. BRCA1 and BRCA2 heterozygosity and repair of X-ray-induced DNA damage. Int. J. Radiat. Biol. 2002, 78, 285–295. [Google Scholar] [CrossRef]
- Boulton, S.J. Cellular functions of the BRCA tumour-suppressor proteins. Biochem. Soc. Trans. 2006, 34, 633–645. [Google Scholar] [CrossRef]
- Andrieu, N.; Easton, D.F.; Chang-Claude, J.; Rookus, M.A.; Brohey, M.R.; Cardis, E.; Antoniou, A.C.; Wagner, T.; Simard, J.; Evans, G.; et al. Effect of chest X-rays on the risk of breast cancer among BRCA1/2 mutation carriers in the international BRCA1/2 carrier cohort study: A report from the EMBRACE, GENEPSO, GEO-HEBON, and IBCCS Collaborators’ Group. J. Clin. Oncol. 2006, 24, 3361–3366. [Google Scholar] [CrossRef] [Green Version]
- Gronwald, J.; Pijpe, A.; Byrski, T.; Huzarski, T.; Stawicka, M.; Cybulski, C.; van Leeuwen, F.; Lubiński, J.; Narod, S.A. Early radiation exposures and BRCA1-associated breast cancer in young women from Poland. Breast Cancer Res. Treat. 2008, 112, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Goldfrank, D.; Chuai, S.; Bernstein, J.L.; Ramon, Y.; Cajal, T.; Lee, J.B.; Alonso, M.C.; Diez, O.; Baiget, M.; Kauff, N.D.; et al. Effect of mammography on breast cancer risk in women with mutations in BRCA1 or BRCA2. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2311–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, N.; Shi, Y.; Yu, L.; Ye, R.; Shan, Z.; Zhang, Z.; Zhang, Y.; Lin, Y. Prospect for Application of PARP Inhibitor in Patients with HER2 Negative Breast Cancer. Int. J. Biol. Sci. 2019, 15, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Nounou, M.I.; ElAmrawy, F.; Ahmed, N.; Abdelraouf, K.; Goda, S.; Syed-Sha-Qhattal, H. Breast Cancer: Conventional Diagnosis and Treatment Modalities and Recent Patents and Technologies. Breast Cancer 2015, 9 (Suppl. 2), 17–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Forker, L.J.; Choudhury, A.; Kiltie, A.E. Biomarkers of Tumour Radiosensitivity and Predicting Benefit from Radiotherapy. Clin. Oncol. 2015, 27, 561–569. [Google Scholar] [CrossRef]
- Yilmaz, M.; Elmali, A.; Yazici, G. Abscopal Effect, from Myth to Reality: From Radiation to Oncologists’ Perspective. Cureus 2019, 11, e3860. [Google Scholar] [CrossRef] [Green Version]
- Pantschenko, A.G.; Pushkar, I.; Anderson, K.H.; Wang, Y.; Miller, L.J.; Kurtzman, S.H.; Kreutzer, D.L. The interleukin-1 family of cytokines and receptors in human breast cancer: Implications for tumor progression. Int. J. Oncol. 2003, 23, 269–284. [Google Scholar] [CrossRef]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Voronov, E. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 2006, 42, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and cancer progression: The emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.S.; Chen, Z.; Dong, G.; Sunwoo, J.B.; Bancroft, C.C.; Capo, D.E.; Van Waes, C. IL (interleukin)-1alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin. Cancer Res. 2001, 7, 1812–1820. [Google Scholar] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.J.; Kurtzman, S.H.; Anderson, K.; Wang, Y.; Stankus, M.; Renna, M.; Kreutzer, D.L. Interleukin-1 family expression in human breast cancer: Interleukin-1 receptor antagonist. Cancer Investig. 2000, 18, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Snoussi, K.; Mahfoudh, W.; Bouaouina, N.; Ahmed, S.B.; Helal, A.N.; Chouchane, L. Genetic variation in IL-8 associated with increased risk and poor prognosis of breast carcinoma. Hum. Immunol. 2006, 67, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zuccari, D.A.; Leonel, C.; Castro, R.; Gelaleti, G.B.; Jardim, B.V.; Moscheta, M.G.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; Neto Dde, S.; et al. An immunohistochemical study of interleukin-8 (IL-8) in breast cancer. Acta Histochem. 2012, 114, 571–576. [Google Scholar] [CrossRef]
- Hamed, E.A.; Zakhary, M.M.; Maximous, D.W. Apoptosis, angiogenesis, inflammation, and oxidative stress: Basic interactions in patients with early and metastatic breast cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 999–1009. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Hall, B.M. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [Green Version]
- Baumgarten, S.C.; Frasor, J. Minireview: Inflammation: An instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol. Endocrinol. 2012, 26, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sanctis, V.; Agolli, L.; Visco, V.; Monaco, F.; Muni, R.; Spagnoli, A.; Campanella, B.; Valeriani, M.; Minniti, G.; Osti, M.F.; et al. Cytokines, fatigue, and cutaneous erythema in early stage breast cancer patients receiving adjuvant radiation therapy. Biomed. Res. Int. 2014, 2014, 523568. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, O.; Yoshiuchi, K.; Inagaki, M.; Matsuoka, Y.; Yoshikawa, E.; Sugawara, Y.; Akechi, T.; Wada, N.; Imoto, S.; Murakami, K.; et al. Association between adjuvant regional radiotherapy and cognitive function in breast cancer patients treated with conservation therapy. Cancer Med. 2014, 3, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sheng, W.; Xiang, J.; Ye, Z.; Zhu, Y.; Chen, X.; Yang, J. Recombinant human IL-24 suppresses lung carcinoma cell growth via induction of cell apoptosis and inhibition of tumor angiogenesis. Cancer Biother. Radiopharm. 2008, 23, 310–320. [Google Scholar] [CrossRef]
- Alagumuthu, M.; Srivastava, V.; Shah, M.; Arumugam, S.K.; Sonaimuthu, M.; Arumugam, N.A. Pro- and Anti-Inflammatory Cytokine Expression Levels in Macrophages; An Approach to Develop Indazolpyridin-Methanones as a Novel Inflammation Medication. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2020, 19. [Google Scholar] [CrossRef]
- Sunpaweravong, S.; Puttawibul, P.; Ruangsin, S.; Laohawiriyakamol, S.; Sunpaweravong, P.; Sangthawan, D.; Pradutkanchana, J.; Raungkhajorn, P.; Geate, A. Randomized Study of Antiinflammatory and ImmuneModulatory Effects of Enteral Immunonutrition during Concurrent Chemoradiotherapy for Esophageal Cancer. Nutr. Cancer 2014, 66, 1–5. [Google Scholar] [CrossRef]
- Wang, H.; Yang, X. Association between serum cytokines and progression of breast cancer in Chinese population. Medicine (Baltimore) 2017, 96, e8840. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Meynköhn, A.; Habermann, N.; Wiskemann, J.; Oelmann, J.; Hof, H.; Wessels, S.; Klassen, O.; Debus, J.; Potthoff, K.; et al. Resistance Exercise and Inflammation in Breast Cancer Patients Undergoing Adjuvant Radiation Therapy: Mediation Analysis from a Randomized, Controlled Intervention Trial. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 329–337. [Google Scholar] [CrossRef]
- Westbury, C.B.; Haviland, J.; Davies, S.; Gothard, L.; Abdi, B.A.; Sydenham, M.; Bowen, J.; Stratton, R.; Short, S.C.; Yarnold, J.R. Cytokine levels as biomarkers of radiation fibrosis in patients treated with breast radiotherapy. Radiat. Oncol. 2014, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.M.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Moses, H.L. Transforming growth factor beta (TGF-β) and inflammation in cancer. Cytokine Growth Factor Rev. 2010, 21, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boothe, D.L.; Coplowitz, S.; Greenwood, E.; Barney, C.L.; Christos, P.J.; Parashar, B.; Nori, D.; Chao, K.S.; Wernicke, A.G. Transforming growth factor β-1 (TGF-β1) is a serum biomarker of radiation induced fibrosis in patients treated with intracavitary accelerated partial breast irradiation: Preliminary results of a prospective study. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 1030–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, S.D.; Bauer, J.; Schmaus, A.; Neumaier, C.; Herskind, C.; Veldwijk, M.R.; Wenz, F.; Sleeman, J.P. TGF-β1 Is Present at High Levels in Wound Fluid from Breast Cancer Patients Immediately Post-Surgery, and Is Not Increased by Intraoperative Radiation Therapy (IORT). PLoS ONE 2016, 11, e0162221. [Google Scholar] [CrossRef] [Green Version]
- Germanov, E.; Berman, J.; Guernsey, D. Current and future approaches for the therapeutic targeting of metastasis. Int. J. Mol. Med. 2006, 18, 2025–2036. [Google Scholar] [CrossRef] [Green Version]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M.S. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010, 112, 3–25. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Melegh, Z.; Oltean, S. Targeting Angiogenesis in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2676. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, S.; Dehghan, M.H.; Dastmalchi, R.; Mashayekhi, F.J.; Salami, S.; Hedayati, M. The roles and role-players in thyroid cancer angiogenesis. Endocr. J. 2019, 66, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The role of vimentin intermediate filaments in the progression of lung cancer. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; De Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, T.; Han, L.; Qin, P.; Wu, Z.; Xu, B.; Gao, Q.; Song, Y. TGFbeta1-induced down-regulation of microRNA-138 contributes to epithelial-mesenchymal transition in primary lung cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Assani, G.; Zhou, Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion (Review). Oncol. Lett. 2019, 17, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef]
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell Biol. 2009, 185, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Hamill, K.J.; Paller, A.S.; Jones, J.C. Adhesion and migration, the diverse functions of the laminin alpha3 subunit. Dermatol. Clin. 2010, 28, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockhorn, M.; Jain, R.K.; Munn, L.L. Active versus passive mechanisms in metastasis: Do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007, 8, 444–448. [Google Scholar] [CrossRef] [Green Version]
- Méhes, G.; Witt, A.; Kubista, E.; Ambros, P.F. Circulating breast cancer cells are frequently apoptotic. Am. J. Pathol. 2001, 159, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Nash, G.F.; Turner, L.F.; Scully, M.F.; Kakkar, A.K. Platelets and Cancer. Lancet Oncol. 2002, 3, 425–430. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, Y.; Lin, E.; Li, J.; Goswami, S.; Stanley, E.; Segall, J.; Pollard, J.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer. 2003, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Wolburg, H. Transendothelial migration of leukocytes: Through the front door or around the side of the house? Eur. J. Immunol. 2004, 34, 2955–2963. [Google Scholar] [CrossRef]
- Tremblay, P.L.; Huot, J.; Auger, F.A. Mechanisms by which E-selectin regulates diapedesis of colon cancer cell under flow conditions. Cancer Res. 2008, 68, 5167–5176. [Google Scholar] [CrossRef] [Green Version]
- Offner, F.A.; Wirtz, H.C.; Schiefer, J.; Bigalke, I.; Klosterhalfen, J.; Bittinger, F.; Mittermayer, C.; Kirkpatrick, C.J. Interactin of human malignant melanoma (ST-ML-12) tumour spheroids with endothelial cell monolayers. Am. J. Pathol. 1992, 141, 601–610. [Google Scholar]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumour formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer. 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; Akslen, L.A.; Folkman, J. Role of angiogenesis in human tumour dormancy: Animal models of the angiogenic switch. Cell Cycle 2006, 5, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Almog, N. Molecular mechanisms underlying tumour dormancy. Cancer Lett. 2010, 294, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Ungar, D. Golgi linked protein glycosylation and associated diseases. Semin. Cell Dev. Biol. 2009, 20, 762–769. [Google Scholar] [CrossRef]
- Sprovieri, P.; Martino, G. The role of the carbohydrates in plasmatic membrane. Physiol. Res. 2018, 67, 1–11. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology, 3rd ed.; Chapters 9 N-Glycans and 10 O-GalNAc Glycans; Publ Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015–2017. [Google Scholar]
- Brooks, S.A. The involvement of Helix pomatia lectin (HPA) binding N-acetylgalactosamine glycans in cancer progression. Histol. Histopathol. 2000, 15, 143–158. [Google Scholar]
- Brooks, S.A. Protein Glycosylation: The Basic Science Chap 2 in Biopharmaceuticals: Post Translational Modifications; Walch, G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 17–49. [Google Scholar]
- Chaze, T.; Slomianny, M.-C.; Milliat, F.; Tarlet, G.; Lefebvre-Darroman, T.; Gourmelon, P.; Bey, E.; Benderitter, M.; Michalski, J.-C.; Guipaud, O. Alteration of the Serum N-glycome of Mice Locally Exposed to High Doses of Ionizing Radiation. Mol. Cell. Proteomics 2013, 12, 283–301. [Google Scholar] [CrossRef] [Green Version]
- Dennis, J.W.; Laferte, S. Oncodevelopmental expression of GlcNac beta 1-6 Man 1-6 Man beta 1-6 branching of Asn-linked oligosaccharides in human breast carcinomas. Cancer Res. 1989, 49, 945–950. [Google Scholar]
- Fernandez, B.; Sagman, U.; Auger, M.; Demetrio, M.; Dennis, J.W. Beta 1-6 branched oligosaccharides as a marker of tumour progression in human breast and colon carcinomas. Cancer Res. 1991, 51, 718–723. [Google Scholar]
- Sears, P.; Wong, C.H. Enzyme action in glycoprotein synthesis. Cell. Mol. Life Sci. 1998, 54, 223–252. [Google Scholar] [CrossRef] [PubMed]
- Hauselmann, I.; Borsig, L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bapu, D.; Runions, J.; Kadhim, M.; Brooks, S.A. N-acetylgalactosamine glycans function in cancer cell adhesion to endothelial cells: A role for truncated O-glycans in metastatic mechanisms. Cancer Lett. 2016, 375, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinsky, V.V.; Glinsky, G.V.; Rittenhouse-Olson, K.; Huflejt, M.E.; Glinskii, O.V.; Deutscher, S.L.; Quinn, T.P. The role of Thomson-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 2001, 61, 4851–4857. [Google Scholar]
- Sewell, R.; Backstrom, M.; Dalziel, M.; Gschmeissner, S.; Karlsson, H.; Noll, T.; Gatgens, J.; Clausen, H.; Hansson, G.C.; Burchell, J.; et al. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J. Biol. Chem. 2006, 281, 3586–3594. [Google Scholar] [CrossRef] [Green Version]
- Tamura, F.; Sato, Y.; Hirakawa, M.; Yoshida, M.; Ono, M.; Osuga, T.; Okagawa, Y.; Uemura, N.; Arihara, Y.; Murase, K.; et al. RNAi-mediated gene silencing of ST6GalNAc I suppresses the metastatic potential in gastric cancer cells. Gastric Cancer 2016, 19, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Sundahl, N.; Duprez, F.; Ost, P.; De Neve, W.; Mareel, M. Effects of radiation on the metastatic process. Mol. Med. 2018, 24, 16. [Google Scholar] [CrossRef] [Green Version]
- Camphausen, K.; Moses, M.A.; Beecken, W.D.; Khan, M.K.; Folkman, J.; O’Reilly, M.S. Radiation therapy to a primary tumor accelerates metastatic growth in mice. Cancer Res. 2001, 61, 2207–2211. [Google Scholar]
- Feys, L.; Descamps, B.; Vanhove, C.; Vral, A.; Veldeman, L.; Vermeulen, S.; De Wagter, C.; Bracke, M.; De Wever, O. Radiation-induced lung damage promotes breast cancer lung-metastasis through CXCR4 signaling. Oncotarget 2015, 6, 26615–26632. [Google Scholar] [CrossRef] [Green Version]
- Kargiotis, O.; Chetty, C.; Gogineni, V.; Gondi, C.S.; Pulukuri, S.M.; Kyritsis, A.P.; Gujrati, M. uPA/uPAR downregulation inhibits radiation-induced migration, invasion and angiogenesis in IOMM-Lee meningioma cells and decreases tumor growth in vivo. Int. J. Oncol. 2008, 33, 937–947. [Google Scholar]
- Kaliski, A.; Maggiorella, L.; Cengel, K.A.; Mathe, D.; Rouffiac, V.; Opolon, P.; Lassau, N.; Bourhis, J.; Deutsch, E. Angiogenesis and tumor growth inhibition by a matrix metalloproteinase inhibitor targeting radiationinduced invasion. Mol. Cancer Ther. 2005, 4, 1717–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi, A.; Griggs, D.W.; Zieher, H.; Roth, A.; Lipson, K.E.; Saffrich, R.; Grone, H.J.; Hallahan, D.E.; Reisfeld, R.A.; Debus, J.; et al. Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin. Cancer Res. 2005, 11, 6270–6279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofia Vala, I.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Ruegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 2010, 5, e11222. [Google Scholar] [CrossRef] [PubMed]
- Kuonen, F.; Laurent, J.; Secondini, C.; Lorusso, G.; Stehle, J.C.; Rausch, T.; Faes-Van’t Hull, E.; Bieler, G.; Alghisi, G.C.; Schwendener, R.; et al. Inhibition of the Kit ligand/c-Kit axis attenuates metastasis in a mouse model mimicking local breast cancer relapse after radiotherapy. Clin. Cancer Res. 2012, 18, 4365–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquette, B.; Baptiste, C.; Therriault, H.; Arguin, G.; Plouffe, B.; Lemay, R. In vitro irradiation of basement membrane enhances the invasiveness of breast cancer cells. Br. J. Cancer 2007, 97, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.W.; Mizumoto, K.; Urashima, T.; Nagai, E.; Maehara, N.; Sato, N.; Nakajima, M.; Tanaka, M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 2002, 8, 1223–1227. [Google Scholar]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor–stromal interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [Green Version]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Lee, H.C.; Kim, M.S.; Lee, S.H.; Park, I.C.; Rhee, C.H.; Hong, S.I. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006, 66, 8511–8519. [Google Scholar] [CrossRef] [Green Version]
- Zhai, G.G.; Malhotra, R.; Delaney, M.; Latham, D.; Nestler, U.; Zhang, M.; Mukherjee, N.; Song, Q.; Robe, P.; Chakravarti, A. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J. Neurooncol. 2006, 76, 227–237. [Google Scholar] [CrossRef]
- Speake, W.J.; Dean, R.A.; Kumar, A.; Morris, T.M.; Scholefield, J.H.; Watson, S.A. Radiation induced MMP expression from rectal cancer is short lived but contributes to in vitro invasion. Eur. J. Surg. Oncol. 2005, 31, 869–874. [Google Scholar] [CrossRef]
- Wang, J.L.; Sun, Y.; Wu, S. Gamma-irradiation induces matrix metalloproteinase II expression in a p53-dependent manner. Mol. Carcinog. 2000, 27, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Young, A.G.H.; Bennewith, K.L. Ionizing radiation enhances breast tumor cell migration in vitro. Radiat. Res. 2017, 188, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Yokoe, T.; Tanaka, K.; Saigusa, S.; Toiyama, Y.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol. Rep. 2012, 27, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Jeon, H.M.; Kim, M.Y.; Kim, C.H.; Park, H.G.; Hans, S.I.; Kang, H.S. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol. Cancer 2017, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, D.; Izumi, S.; Suzuki, F.; Kamiya, K. Analysis of a lectin microarray identifies altered sialylation of mouse serum glycoproteins induced by whole-body radiation exposure. J. Radiat. Res. 2019, 60, 189–196. [Google Scholar] [CrossRef]
- Toth, E.; Vekey, K.; Ozohanics, O.; Jeko, A.; Dominczyk, I.; Widlak, P.; Drahos, L. Changes of protein glycosylation in the course of radiotherapy. J. Pharm. Biomed. Anal. 2016, 118, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Jaillet, C.; Morelle, W.; Slomianny, M.-C.; Paget, V.; Tarlet, G.; Buard, V.; Selbonne, S.; Caffin, F.; Emilie Rannou, E.; Martinez, P.; et al. Radiation-induced changes in the glycome of endothelial cells with functional consequences. Sci. Rep. 2017, 7, 5290. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-B.; Shen, L.; Qui, L. Reversal effect of GnT-V on the radioresistance of human nasopharangeal carcinoma cells by alteration beta 1,6-GlcNAc branched N-glycans. Int. J. Clin. Exp. Pathol. 2015, 8, 9901–9911. [Google Scholar]
- Shen, L.; Dong, X.-X.; Wu, J.-B. Radiosensitisation of human glioma cells by inhibition of beta 1,6-GlcNAc branched N-glycans. Tumor Biol. 2016, 37, 4909–4918. [Google Scholar] [CrossRef]
- Rutqvist, L.E.; Rose, C.; Cavallin-stahl, E. A systematic overview of radiation therapy effects in breast cancer. Acta Ocol. 2003, 42, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Budach, W.; Kammers, K.; Boelke, E.; Matuschek, C. Adjuvant radiotherapy of regional lymph nodes in breast cancer—A meta-analysis of randomised trials. Radiat. Oncol. 2013, 8, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, O.A.; Anderson, R.L.; Russell, P.A.; Cox, R.A.; Ivashkevich, A.; Swierczak, A.; Doherty, J.P.; Jacobs, D.H.; Smith, J.; Siva, S.; et al. Mobilization of viable tumor cells into the circulation during radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, J.F.; Kao, G.D.; MacArthur, K.M.; Ju, M.; Steinmetz, D.; Wileyto, E.P.; Simone, C.B., II; Hahn, S.M. Tracking viable circulating tumor cells (CTCs) in the peripheral blood of non-small cell lung cancer patients undergoing definitive radiation therapy: Pilot study results. Cancer 2015, 121, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Lowes, L.E.; Lock, M.; Rodrigues, G.; D’Souza, D.; Bauman, G.; Ahmad, B.; Ventkatesan, V.; Allan, A.L.; Sexton, T. Circulating tumour cells in prostate cancer patients receiving salvage radiotherapy. Clin. Transl. Oncol. 2012, 14, 150–156. [Google Scholar] [CrossRef]
- Couto, N.; Caja, S.; Maia, J.; Strano Moraes, M.C.; Costa-Silva, B. Exosomes as emerging players in cancer biology. Biochimie 2018, 155, 2–10. [Google Scholar] [CrossRef]
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thêry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef]
- Ge, R.; Tan, E.; Sharghi-Namini, S.; Asada, H.H. Exosomes in Cancer Microenvironment and Beyond: Have we Overlooked these Extracellular Messengers? Cancer Microenviron. 2012, 5, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Jella, K.K.; Rani, S.; O’Driscoll, L.; McClean, B.; Byrne, H.J.; Lyng, F.M. Exosomes are involved in mediating radiation induced bystander signaling in human keratinocyte cells. Radiat. Res. 2014, 181, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.; Fatima, F.; Nazarenko, I.; Ekström, K.; Murtaza, I.; Anees, M.; Sultan, A.; Neder, L.; Camussi, G.; Valadi, H. Extracellular vesicles in ovarian cancer: Applications to tumor biology, immunotherapy and biomarker discovery. Expert Rev. Proteomic 2016, 13, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Fatima, F.; Nawaz, M. Vesiculated long non-coding rnas: Offshore packages deciphering trans-regulation between cells, cancer progression and resistance to therapies. Non-Coding RNA 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arscott, W.T.; Tandle, A.T.; Zhao, S.; Shabason, J.E.; Gordon, I.K.; Schlaff, C.D.; Zhang, G.; Tofilon, P.J.; Camphausen, K.A. Ionizing radiation and glioblastoma exosomes: Implications in tumor biology and cell migration. Transl. Oncol. 2013, 6, 638–648. [Google Scholar] [CrossRef] [Green Version]
- Xin, T.; Greco, V.; Myung, P. Hardwiring stem cell communication through tissue structure. Cell 2016, 164, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Steinbichler, T.B.; Dudas, J.; Riechelmann, H.; Skvortsova, I.I. The role of exosomes in cancer metastasis. Semin. Cancer Biol. 2017, 44, 170–181. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Organotropic metastasis: Role of tumor exosomes. Cell Res. 2016, 26, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, J.Q.; Liu, J.L.; Tian, L. Exosomes in tumor microenvironment: Novel transporters and biomarkers. J. Transl. Med. 2016, 14, 297. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.; Sugimoto, H.; O’Connell, J.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.; Melo, C.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dioufa, N.; Clark, A.; Ma, B.; Beckwitt, C.; Wells, A. Bi-directional exosome-driven intercommunication between the hepatic niche and cancer cells. Mol. Cancer 2017, 16, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Battafarano, G.; D’Agostini, M.; Del Fattore, A. The Role of Extracellular Vesicles in Bone Metastasis. Int. J. Mol. Sci. 2018, 19, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, S.; Israel, S.; Nagy, C.; Turecki, G. The emerging role of exosomes in mental disorders. Transl. Psychiatry 2019, 9, 122. [Google Scholar] [CrossRef]
- Abramowicz, A.; Wojakowska, A.; Marczak, L.; Lysek-Gladysinska, M.; Smolarz, M.; Story, M.; Polanska, J.; Widlak, P.; Pietrowska, M. Ionizing radiation affects the composition of the proteome of extracellular vesicles released by head-and-neck cancer cells in vitro. J. Radiat. Res. 2019, 60, 289–297. [Google Scholar] [CrossRef]
- Jelonek, K.; Wojakowska, A.; Marczak, L.; Muer, A.; Tinhofer-Keilholz, I.; Lysek-Gladysinska, M.; Widlak, P.; Pietrowska, M. Ionizing radiation affects protein composition of exosomes secreted in vitro from head and neck squamous cell carcinoma. Acta Biochim. Pol. 2015, 62, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Al-Mayah, A.; Bright, S.; Chapman, K.; Irons, S.; Luo, P.; Carter, D.; Goodwin, E.; Kadhim, M. The non-targeted effects of radiation are perpetuated by exosomes. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 772, 38–45. [Google Scholar] [CrossRef]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle trex1-sensitive ifn-stimulatory dsdna from irradiated cancer cells to dcs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Al-Mayah, A.H.; Bright, S.J.; Bowler, D.A.; Slijepcevic, P.; Goodwin, E.; Kadhim, M.A. Exosome Mediated Telomere Instability in Human Breast Epithelial Cancer Cells after X Irradiation. Radiat. Res. 2017, 187, 98–106. [Google Scholar] [CrossRef]
- Wen, S.; Sceneay, J.; Lima, L.; Wong, C.; Becker, M.; Krumeich, S.; Lobb, R.; Castillo, V.; Wong, K.; Ellis, S.; et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Wan, T.; Wang, B.; Zhou, X.; Xiu, F.; Chen, T.; Wu, Y.; Cao, X. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mayah, A.H.; Irons, S.L.; Pink, R.C.; Carter, D.R.; Kadhim, M.A. Possible role of exosomes containing RNA in mediating nontargeted effect of ionizing radiation. Radiat. Res. 2012, 177, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freudenmann, L.K.; Mayer, C.; Rodemann, H.P.; Dittmann, K. Reduced exosomal L-Plastin is responsible for radiation-induced bystander effect. Exp. Cell Res. 2019, 383, 111498. [Google Scholar] [CrossRef]
- De Araujo Farias, V.; O’Valle, F.; Serrano-Saenz, S.; Anderson, P.; Andrés, E.; López Peñalver, J.; Tovar, I.; Nieto, A.; Santos, A.; Martín, F.; et al. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol. Cancer 2018, 17, 122. [Google Scholar] [CrossRef]
- Tosetti, F.; Venè, R.; Camodeca, C.; Nuti, E.; Rossello, A.; D’Arrigo, C.; Galante, D.; Ferrari, N.; Poggi, A.; Zocchi, M.R. Specific ADAM10 inhibitors localize in exosome-like vesicles released by Hodgkin lymphoma and stromal cells and prevent sheddase activity carried to bystander cells. Oncoimmunology 2018, 7, e1421889. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadhim, M.A.; Mayah, A.; Brooks, S.A. Does Direct and Indirect Exposure to Ionising Radiation Influence the Metastatic Potential of Breast Cancer Cells. Cancers 2020, 12, 236. https://doi.org/10.3390/cancers12010236
Kadhim MA, Mayah A, Brooks SA. Does Direct and Indirect Exposure to Ionising Radiation Influence the Metastatic Potential of Breast Cancer Cells. Cancers. 2020; 12(1):236. https://doi.org/10.3390/cancers12010236
Chicago/Turabian StyleKadhim, Munira A., Ammar Mayah, and Susan A. Brooks. 2020. "Does Direct and Indirect Exposure to Ionising Radiation Influence the Metastatic Potential of Breast Cancer Cells" Cancers 12, no. 1: 236. https://doi.org/10.3390/cancers12010236
APA StyleKadhim, M. A., Mayah, A., & Brooks, S. A. (2020). Does Direct and Indirect Exposure to Ionising Radiation Influence the Metastatic Potential of Breast Cancer Cells. Cancers, 12(1), 236. https://doi.org/10.3390/cancers12010236