Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity

 , and
, and
Abstract
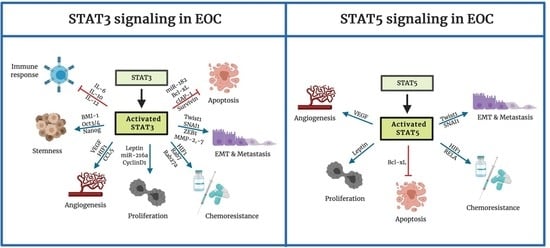
:
1. Introduction
2. Regulation of STAT3/STAT5 Activation in EOC
2.1. IL-6 Pathway
2.2. TP53 Mutation
2.3. Lipid Metabolism Pathway
2.4. Receptor Tyrosine Kinases (RTKs)
2.5. Alternative Cytokine or Non-Receptor Tyrosine Kinases
2.6. Other Gene Regulatory Mechanisms
3. The Function of STAT3 and STAT5 in EOC
3.1. Apoptosis
3.2. Proliferation
3.3. Angiogenesis
3.4. Tumor Progression and Metastasis
4. Tumor Microenvironment
4.1. Non-Immune Stromal Cells
4.2. Immune Function
4.3. Tumor-Associated Macrophages
5. Targeting of the STAT3/STAT5 Signaling Pathway in EOC
6. Conclusions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Mediators | Effect | References |
|---|---|---|
| miR-17-5b | increase | [163] |
| miR-133b | increase | [164] |
| miR-134 | increase | [165] |
| miR-135a | increase | [164] |
| miR-147 | increase | [166] |
| miR-182 | increase | [167] |
| miR-204 | increase | [168] |
| miR-874 | increase | [169] |
| Bcl-xL | increase | [57,170,171] |
| Bcl-2 | increase | [52,57,172] |
| cIAP-1 | increase | [173] |
| Mcl-1L | increase | [172,174,175] |
| Fas | decrease | [171,176] |
| Survivin | increase | [57] |
| Mediators | Effect | References |
|---|---|---|
| VEGF | increase | [80,177,178,179] |
| HIF1 | increase | [76,159,180,181] |
| CCL5 | increase | [111,117] |
| Mediators | Effect | References |
|---|---|---|
| MMP-2 | increase | [182,183] |
| MMP-7 | increase | [184] |
| MMP-9 | increase | [103] |
| Mediators | Effect | References |
|---|---|---|
| Twist1 | increase | [88,185,186,187,188] |
| Snail1 | increase | [87,186,189] |
| Zeb | increase | [86] |
| miR-200 family | decrease | [86] |
| let7 | decrease | [86] |
References
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A. Histologic Subtypes of Ovarian Carcinoma. Int. J. Gynecol. Pathol. 2008, 27, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Bookman, M.A.; Brady, M.F.; McGuire, W.P.; Harper, P.G.; Alberts, D.S.; Friedlander, M.; Colombo, N.; Fowler, J.M.; Argenta, P.A.; De Geest, K.; et al. Evaluation of New Platinum-Based Treatment Regimens in Advanced-Stage Ovarian Cancer: A Phase III Trial of the Gynecologic Cancer InterGroup. J. Clin. Oncol. 2009, 27, 1419–1425. [Google Scholar] [CrossRef]
- Baldwin, L.A.; Miller, R.W.; Tucker, T.; Goodrich, S.T.; Podzielinski, I.; Ueland, F.R.; Seamon, L.G.; Huang, B.; DeSimone, C.P.; Van Nagell, J.R. Ten-Year Relative Survival for Epithelial Ovarian Cancer. Obstet. Gynecol. 2012, 120, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Signalling: STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, e651. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, E.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Teglund, S.; McKay, C.; Schuetz, E.; Van Deursen, J.M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G.; Ihle, J.N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 1998, 93, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer-New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Burke, W.M.; Jin, X.; Lin, H.-J.; Huang, M.; Liu, R.; Reynolds, R.K.; Lin, J. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 2001, 20, 7925–7934. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Page, C.; Reynolds, R.; Lin, J. Constitutive Activation of Stat 3 Oncogene Product in Human Ovarian Carcinoma Cells. Gynecol. Oncol. 2000, 79, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Savarese, T.M.; Campbell, C.L.; McQuain, C.; Mitchell, K.; Guardiani, R.; Quesenberry, P.J.; Nelson, B.E. Coexpression of oncostatin m and its receptors and evidence for stat3 activation in human ovarian carcinomas. Cytokine 2002, 17, e324. [Google Scholar] [CrossRef]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (STAT) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, e3550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ye, D.; Xie, X.; Chen, B.; Lü, W. VEGF, VEGFRs expressions and activated STATs in ovarian epithelial carcinoma. Gynecol. Oncol. 2004, 94, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Wei-Hong, Z. Constitutive activation of signal transducer and activator of transcription 3 in epithelial ovarian carcinoma. J. Obstet. Gynaecol. Res. 2009, 35, 918–925. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Miyamoto, M.; Aoyama, T.; Soyama, H.; Goto, T.; Hirata, J.; Suzuki, A.; Nagaoka, I.; Tsuda, H.; Furuya, K.; et al. JAK2/STAT3 pathway as a therapeutic target in ovarian cancers. Oncol. Lett. 2018, 15, 5772–5780. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.G.; Yang, G.; Bast, R.C.; Amin, H.M.; Lai, R.; Liu, J.; Mercado-Uribe, I. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer 2006, 107, 2730–2740. [Google Scholar] [CrossRef]
- Chen, M.-W.; Yang, S.-T.; Chien, M.-H.; Wu, C.-J.; Hua, K.-T.; Hsiao, S.; Lin, H.; Su, J.-L.; Wei, L.-H. The STAT3-miRNA-92-Wnt Signaling Pathway Regulates Spheroid Formation and Malignant Progression in Ovarian Cancer. Cancer Res. 2017, 77, 1955–1967. [Google Scholar] [CrossRef] [Green Version]
- Jinawath, N.; Vasoontara, C.; Jinawath, A.; Fang, X.; Zhao, K.; Yap, K.-L.; Guo, T.; Lee, C.S.; Wang, W.; Balgley, B.M.; et al. Oncoproteomic Analysis Reveals Co-Upregulation of RELA and STAT5 in Carboplatin Resistant Ovarian Carcinoma. PLoS ONE 2010, 5, e11198. [Google Scholar] [CrossRef]
- Koskela, H.L.; Eldfors, S.; Ellonen, P.; Van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, R.; Ma, L.; Merker, J.; Martinez, B.; Zehnder, J.; Arber, D. STAT3 mutations are frequent in CD30+ T-cell lymphomas and T-cell large granular lymphocytic leukemia. Leukemia 2013, 27, e2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, H.L.M.; Eldfors, S.; Kuusanmäki, H.; Van Adrichem, A.J.; Olson, T.; Lagström, S.; Andersson, E.I.; Jerez, A.; Clemente, M.J.; Yan, Y.; et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 2013, 121, 4541–4550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Hibi, M.; Nakagawa, N.; Nakagawa, T.; Yasukawa, K.; Yamanishi, K.; Taga, T.; Kishimoto, T. IL-6-induced homodimerization of gp130 and associated activation of a tyrosine kinase. Science 1993, 260, 1808–1810. [Google Scholar] [CrossRef]
- Kumar, J.; Ward, A.C. Role of the interleukin 6 receptor family in epithelial ovarian cancer and its clinical implications. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1845, 117–125. [Google Scholar] [CrossRef]
- McLean, K.; Tan, L.; Bolland, D.E.; Coffman, L.G.; Peterson, L.F.; Talpaz, M.; Neamati, N.; Buckanovich, R.J. Leukemia inhibitory factor functions in parallel with interleukin-6 to promote ovarian cancer growth. Oncogene 2019, 38, e1576. [Google Scholar] [CrossRef]
- Zhou, W.; Sun, W.; Yung, M.M.H.; Dai, S.; Cai, Y.; Chen, C.-W.; Meng, Y.; Lee, J.B.; Braisted, J.C.; Xu, Y.; et al. Autocrine activation of JAK2 by IL-11 promotes platinum drug resistance. Oncogene 2018, 37, 3981–3997. [Google Scholar] [CrossRef]
- Reid, T.; Jin, X.; Song, H.; Tang, H.-J.; Reynolds, R.K.; Lin, J. Modulation of Janus kinase 2 by p53 in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2004, 321, 441–447. [Google Scholar] [CrossRef]
- Pavelka, J.C.; Brown, R.S.; Karlan, B.Y.; Cass, I.; Leuchter, R.S.; Lagasse, L.D.; Li, A.J. Effect of obesity on survival in epithelial ovarian cancer. Cancer 2006, 107, 1520–1524. [Google Scholar] [CrossRef]
- Seo, J.-M.; Park, S.; Kim, J.-H. Leukotriene B4 Receptor-2 Promotes Invasiveness and Metastasis of Ovarian Cancer Cells through Signal Transducer and Activator of Transcription 3 (STAT3)-dependent Up-regulation of Matrix Metalloproteinase 2*. J. Boil. Chem. 2012, 287, 13840–13849. [Google Scholar] [CrossRef] [Green Version]
- Uddin, S.; Bu, R.; Ahmed, M.; Abubaker, J.; Al-Dayel, F.; Bavi, P.; Al-Kuraya, K.S. Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol. Cancer 2009, 8, e74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-H.; Lee, K.-T.; Leung, P.C. Estrogen receptor alpha pathway is involved in leptin-induced ovarian cancer cell growth. Carcinogenesis 2011, 32, e589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Park, G.; Kim, D. MicroRNA-503-5p Inhibits the CD97-Mediated JAK2/STAT3 Pathway in Metastatic or Paclitaxel-Resistant Ovarian Cancer Cells. Neoplasia 2019, 21, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Chen, H.; Yel, F.; Wang, F.; Xie, X. VEGF induces phosphorylation of STAT3 through binding VEGFR2 in ovarian carcinoma cells in vitro. Eur. J. Gynaecol. Oncol. 2006, 27, 363–369. [Google Scholar] [PubMed]
- Colomiere, M.; Ward, A.; Riley, C.; Trenerry, M.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial–mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2009, 100, e134. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Dobrzycka, B.; Terlikowski, S.J.; Kowalczuk, O.; Niklińska, W.; Chyczewski, L.; Kulikowski, M. Mutations in the KRAS gene in ovarian tumors. Folia Histochem. Cytobiol. 2009, 47, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yue, P.; Zhang, X.; Paladino, D.; Sengupta, B.; Ahmad, S.; Holloway, R.W.; Ingersoll, S.B.; Turkson, J. Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene 2011, 31, 2309–2322. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.; Fraser, F.; Riley, C.; Ahmed, N.; McCulloch, D.; Ward, A. Granulocyte colony-stimulating factor receptor signalling via Janus kinase 2/signal transducer and activator of transcription 3 in ovarian cancer. Br. J. Cancer 2014, 110, e133. [Google Scholar] [CrossRef]
- Guo, Q.; Lu, L.; Liao, Y.; Wang, X.; Zhang, Y.; Liu, Y.; Huang, S.; Sun, H.; Li, Z.; Zhao, L. Influence of c-Src on hypoxic resistance to paclitaxel in human ovarian cancer cells and reversal of FV-429. Cell Death Dis. 2018, 8, e3178. [Google Scholar] [CrossRef] [Green Version]
- Pradeep, S.; Huang, J.; Mora, E.M.; Nick, A.M.; Cho, M.S.; Wu, S.Y.; Noh, K.; Pecot, C.V.; Rupaimoole, R.; Stein, M.A.; et al. Erythropoietin Stimulates Tumor Growth via EphB4. Cancer Cell 2015, 28, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aapro, M.; Jelkmann, W.; Constantinescu, S.N.; Leyland-Jones, B. Effects of erythropoietin receptors and erythropoiesis-stimulating agents on disease progression in cancer. Br. J. Cancer 2012, 106, 1249–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaluvally-Raghavan, P.; Jeong, K.J.; Pradeep, S.; Silva, A.M.; Yu, S.; Liu, W.; Moss, T.; Rodriguez-Aguayo, C.; Zhang, N.; Ram, P.; et al. Direct Upregulation of STAT3 by MicroRNA-551b-3p Deregulates Growth and Metastasis of Ovarian Cancer. Cell Rep. 2016, 15, 1493–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Boil. Ther. 2010, 10, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Cai, Q.; Godwin, A.K.; Zhang, R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol. Cancer Res. 2010, 8, 1610–1618. [Google Scholar] [CrossRef] [Green Version]
- Özeş, A.R.; Pulliam, N.; Ertosun, M.G.; Yılmaz, Ö.; Tang, J.; Çopuroğlu, E.; Matei, D.; Ozes, O.N.; Nephew, K.P. Protein kinase A-mediated phosphorylation regulates STAT3 activation and oncogenic EZH2 activity. Oncogene 2018, 37, 3589–3600. [Google Scholar] [CrossRef]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.-H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.-B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Mandal, M.; Powers, S.E.; Maienschein-Cline, M.; Bartom, E.T.; Hamel, K.M.; Kee, B.L.; Dinner, A.R.; Clark, M.R. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat. Immunol. 2011, 12, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Orlova, A.; Wagner, C.; De Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserű, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, e2. [Google Scholar] [CrossRef] [PubMed]
- Bixel, K.; Saini, U.; Bid, H.K.; Fowler, J.; Riley, M.; Wanner, R.; Dorayappan, K.D.P.; Rajendran, S.; Konishi, I.; Matsumura, N.; et al. Targeting STAT3 by HO3867 induces apoptosis in ovarian clear cell carcinoma. Int. J. Cancer 2017, 141, 1856–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Fang, H.; McCulloch, D.R.; Crowley, T.; Ward, A.C. Leptin receptor signaling via Janus kinase 2/Signal transducer and activator of transcription 3 impacts on ovarian cancer cell phenotypes. Oncotarget 2017, 8, 93530–93540. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.P.; Raaijmakers, J.A.; Lammers, J.-W.J.; Koenderman, L. STAT5-Dependent CyclinD1 and Bcl-xL Expression in Bcr-Abl-Transformed Cells. Mol. Cell Boil. Res. Commun. 2000, 3, 299–305. [Google Scholar] [CrossRef]
- Sánchez-Ceja, S.; Reyes-Maldonado, E.; Vázquez-Manríquez, M.; López-Luna, J.; Belmont, A.; Gutiérrez-Castellanos, S. Differential expression of STAT5 and Bcl-xL, and high expression of Neu and STAT3 in non-small-cell lung carcinoma. Lung Cancer 2006, 54, 163–168. [Google Scholar] [CrossRef]
- Guo, Y.; Nemeth, J.; O’Brien, C.; Susa, M.; Liu, X.; Zhang, Z.; Choy, E.; Mankin, H.; Hornicek, F.; Duan, Z. Effects of Siltuximab on the IL-6-Induced Signaling Pathway in Ovarian Cancer. Clin. Cancer Res. 2010, 16, 5759–5769. [Google Scholar] [CrossRef] [Green Version]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Li, F.; et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef]
- Tian, F.; Jia, L.; Chu, Z.; Han, H.; Zhang, Y.; Cai, J. MicroRNA-519a inhibits the proliferation and promotes the apoptosis of ovarian cancer cells through targeting signal transducer and activator of transcription 3. Exp. Ther. Med. 2017, 15, 1819–1824. [Google Scholar] [CrossRef] [Green Version]
- Syed, V. Reproductive Hormone-Induced, STAT3-Mediated Interleukin 6 Action in Normal and Malignant Human Ovarian Surface Epithelial Cells. J. Natl. Cancer Inst. 2002, 94, 617–629. [Google Scholar] [CrossRef]
- Nagle, C.M.; Australian Ovarian Cancer Study Group; Dixon, S.C.; Jensen, A.; Kjaer, S.K.; Modugno, F.; DeFazio, A.; Fereday, S.; Hung, J.; Johnatty, S.E.; et al. Obesity and survival among women with ovarian cancer: Results from the Ovarian Cancer Association Consortium. Br. J. Cancer 2015, 113, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.-T.; Wang, L.-M.; Hsieh, M.-T.; Shih, Y.-J.; Nana, A.W.; Changou, C.A.; Yang, Y.-C.S.H.; Chiu, H.-C.; Fu, E.; Davis, P.J.; et al. Leptin OB3 peptide suppresses leptin-induced signaling and progression in ovarian cancer cells. J. Biomed. Sci. 2017, 24, e51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Dong, Z.; Li, Y.; Yang, Y.; Yuan, Z.; Qu, X.; Kong, B. The upregulation of signal transducer and activator of transcription 5-dependent microRNA-182 and microRNA-96 promotes ovarian cancer cell proliferation by targeting forkhead box O3 upon leptin stimulation. Int. J. Biochem. Cell Boil. 2013, 45, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.E.; Walker, A.M. Prolactin inhibits a major tumor-suppressive function of wild type BRCA1. Cancer Lett. 2016, 375, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedor, E.; Gregoraszczuk, E. Łucja the molecular mechanism of action of superactive human leptin antagonist (SHLA) and quadruple leptin mutein Lan-2 on human ovarian epithelial cell lines. Cancer Chemother. Pharmacol. 2016, 78, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Kuppusamy, M.L.; Ahmed, S.; Bratasz, A.; Meenakshisundaram, G.; Rivera, B.K.; Khan, M.; Kuppusamy, P. Oxygenation inhibits ovarian tumor growth by downregulating STAT3 and cyclin-D1 expressions. Cancer Boil. Ther. 2010, 10, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Liu, Y.; Wang, R. STAT3 regulated miR-216a promotes ovarian cancer proliferation and cisplatin resistance. Biosci. Rep. 2018, 38, BSR20180547. [Google Scholar] [CrossRef]
- Jiang, Q.; Dai, L.; Cheng, L.; Chen, X.; Li, Y.; Zhang, S.; Su, X.; Zhao, X.; Wei, Y.; Deng, H. Efficient inhibition of intraperitoneal ovarian cancer growth in nude mice by liposomal delivery of short hairpin RNA against STAT3. J. Obstet. Gynaecol. Res. 2013, 39, 701–709. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, L.; Zhang, X.; Liu, R. Synergistic effects of eukaryotic co-expression plasmid-based STAT3-specific siRNA and LKB1 on ovarian cancer in vitro and in vivo. Oncol. Rep. 2015, 33, e774. [Google Scholar] [CrossRef]
- Zhao, S.-H.; Zhao, F.; Zheng, J.-Y.; Gao, L.-F.; Zhao, X.-J.; Cui, M.-H. Knockdown of stat3 expression by RNAi inhibits in vitro growth of human ovarian cancer. Radiol. Oncol. 2011, 45, 196–203. [Google Scholar] [CrossRef]
- Saydmohammed, M.; Joseph, D.; Syed, V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in ovarian and endometrial cancer cells. J. Cell. Biochem. 2010, 110, 447. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Lowe, G.; Roberts, C.M.; Finlay, J.; Han, E.S.; Glackin, C.A.; Dellinger, T.H. Pterostilbene Suppresses Ovarian Cancer Growth via Induction of Apoptosis and Blockade of Cell Cycle Progression Involving Inhibition of the STAT3 Pathway. Int. J. Mol. Sci. 2018, 19, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Cao, Y.; Chen, L.; Liu, F.; Qi, Z.; Cheng, X.; Wang, Z. Cryptotanshinone suppresses cell proliferation and glucose metabolism via STAT3/SIRT3 signaling pathway in ovarian cancer cells. Cancer Med. 2018, 7, 4610–4618. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Wang, L.; Yao, J.C.; Ajani, J.A.; Wei, D.; Aldape, K.D.; Xie, K.; Sawaya, R.; Huang, S. Expression of Activated Signal Transducer and Activator of Transcription 3 Predicts Expression of Vascular Endothelial Growth Factor in and Angiogenic Phenotype of Human Gastric Cancer. Clin. Cancer Res. 2005, 11, 1386–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, C.; Ruemmele, P.; Gehmert, S.; Schenk, H.; Kreutz, M.P.; Mycielska, M.E.; Hackl, C.; Kroemer, A.; ASchnitzbauer, A.; Stoeltzing, O.; et al. STAT5b as Molecular Target in Pancreatic Cancer-Inhibition of Tumor Growth, Angiogenesis, and Metastases. Neoplasia 2012, 14, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Anglesio, M.S.; George, J.; Kulbe, H.; Friedlander, M.; Rischin, D.; Lemech, C.; Power, J.; Coward, J.; Cowin, P.A.; House, C.M.; et al. IL6-STAT3-HIF Signaling and Therapeutic Response to the Angiogenesis Inhibitor Sunitinib in Ovarian Clear Cell Cancer. Clin. Cancer Res. 2011, 17, 2538–2548. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.-H.; Kuo, M.-L.; Chen, C.-A.; Chou, C.-H.; Lai, K.-B.; Lee, C.-N.; Hsieh, C.-Y. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Han, E.S.; Dellinger, T.H.; Lu, J.; Nam, S.; Anderson, R.A.; Yim, J.H.; Wen, W. Cinnamon extract reduces VEGF expression via suppressing HIF-1α gene expression and inhibits tumor growth in mice. Mol. Carcinog. 2017, 56, 436–446. [Google Scholar] [CrossRef]
- Kandala, P.K.; Srivastava, S.K. Regulation of Janus-activated kinase-2 (JAK2) by diindolylmethane in ovarian cancer in vitro and in vivo. Drug Discov. Ther. 2012, 6, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Eichten, A.; Su, J.; Adler, A.P.; Zhang, L.; Ioffe, E.; Parveen, A.A.; Yancopoulos, G.D.; Rudge, J.S.; Lowy, I.; Lin, H.C.; et al. Resistance to Anti-VEGF Therapy Mediated by Autocrine IL6/STAT3 Signaling and Overcome by IL6 Blockade. Cancer Res. 2016, 76, 2327–2339. [Google Scholar] [CrossRef] [Green Version]
- Güth, U.; Huang, D.J.; Bauer, G.; Stieger, M.; Wight, E.; Singer, G. Metastatic patterns at autopsy in patients with ovarian carcinoma. Cancer 2007, 110, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Geng, L.; Zeng, L.; Liu, F.; Huang, Q. AKT2 contributes to increase ovarian cancer cell migration and invasion through the AKT2-PKM2-STAT3/NF-κB axis. Cell. Signal. 2018, 45, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, A.M.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, e128. [Google Scholar] [CrossRef] [PubMed]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C.R. Epithelial to Mesenchymal Transition Promotes Breast Cancer Progression via a Fibronectin-dependent STAT3 Signaling Pathway*. J. Boil. Chem. 2013, 288, 17954–17967. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28–let-7–HMGA2 and miR-200–ZEB1 circuits initiate and maintain oncostatin M-driven epithelial–mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Kim, M.-J.; Lim, J.; Yang, Y.; Lee, M.-S.; Lim, J.-S. N-myc downstream-regulated gene 2 (NDRG2) suppresses the epithelial–mesenchymal transition (EMT) in breast cancer cells via STAT3/Snail signaling. Cancer Lett. 2014, 354, 33–42. [Google Scholar] [CrossRef]
- Chong, K.Y.; Kang, M.; Garofalo, F.; Ueno, D.; Liang, H.; Cady, S.; Madarikan, O.; Pitruzzello, N.; Tsai, C.-H.; Hartwich, T.M.; et al. Inhibition of Heat Shock Protein 90 suppresses TWIST1 Transcription. Mol. Pharmacol. 2019, 96, 168–179. [Google Scholar] [CrossRef]
- Abubaker, K.; Luwor, R.B.; Escalona, R.; McNally, O.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Targeted Disruption of the JAK2/STAT3 Pathway in Combination with Systemic Administration of Paclitaxel Inhibits the Priming of Ovarian Cancer Stem Cells Leading to a Reduced Tumor Burden. Front. Oncol. 2014, 4, e75. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Gwak, H.; Kim, H.S.; Kim, B.; Dhanasekaran, D.N.; Song, Y.S. Malignant ascites enhances migratory and invasive properties of ovarian cancer cells with membrane bound IL-6R in vitro. Oncotarget 2016, 7, 83148–83159. [Google Scholar] [CrossRef] [Green Version]
- Saini, U.; Naidu, S.; ElNaggar, A.; Bid, H.; Wallbillich, J.; Bixel, K.; Bolyard, C.; Suarez, A.; Kaur, B.; Kuppusamy, P.; et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: A potential therapeutic target. Oncogene 2017, 36, e168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and Characterization of Tumor Cells from the Ascites of Ovarian Cancer Patients: Molecular Phenotype of Chemoresistant Ovarian Tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.-H.; Jia, Y.; Guo, F.-J.; Chen, J.; Zhang, X.-W.; Cui, M.-H. Phosphorylation of STAT3 at Tyr705 regulates MMP-9 production in epithelial ovarian cancer. PLoS ONE 2017, 12, e0183622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Lin, Y.G.; Armaiz-Pena, G.N.; Das, P.D.; Arevalo, J.M.; Kamat, A.A.; Han, L.Y.; Jennings, N.B.; Spannuth, W.A.; Thaker, P.H.; et al. Neuroendocrine Modulation of Signal Transducer and Activator of Transcription-3 in Ovarian Cancer. Cancer Res. 2007, 67, 10389–10396. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Zhang, X.; Xu, C. IL6-induced metastasis modulators p-STAT3, MMP-2 and MMP-9 are targets of 3,3’-diindolylmethane in ovarian cancer cells. Cell Oncol. 2016, 39, e47. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Boil. Chem. 2008, 283, 17635–17651. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sun, J.; Cai, B.; Xi, X.; Yang, L.; Zhang, Z.; Feng, Y.; Sun, Y. NANOG regulates epithelial-mesenchymal transition and chemoresistance through activation of the STAT3 pathway in epithelial ovarian cancer. Tumor Boil. 2016, 37, 9671–9680. [Google Scholar] [CrossRef]
- Liu, A.; Fan, D.; Wang, Y. The BET bromodomain inhibitor i-BET151 impairs ovarian cancer metastasis and improves antitumor immunity. Cell Tissue Res. 2018, 374, 577–585. [Google Scholar] [CrossRef]
- Yanaihara, N.; Hirata, Y.; Yamaguchi, N.; Noguchi, Y.; Saito, M.; Nagata, C.; Takakura, S.; Yamada, K.; Okamoto, A. Antitumor effects of interleukin-6 (IL-6)/interleukin-6 receptor (IL-6R) signaling pathway inhibition in clear cell carcinoma of the ovary. Mol. Carcinog. 2015, 55, 832–841. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wallbillich, J.J.; Cohn, D.E.; Selvendiran, K. The biological significance and clinical applications of exosomes in ovarian cancer. Gynecol. Oncol. 2016, 142, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhu, X.; Fei, J.; Shi, P.; Yu, S.; Zhou, J. Advances of exosome in the development of ovarian cancer and its diagnostic and therapeutic prospect. OncoTargets Ther. 2018, 11, 2831–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Sawada, K.; Kinose, Y.; Yoshimura, A.; Toda, A.; Nakatsuka, E.; Hashimoto, K.; Mabuchi, S.; Morishige, K.; Kurachi, H.; et al. Exosomes Promote Ovarian Cancer Cell Invasion through Transfer of CD44 to Peritoneal Mesothelial Cells. Mol. Cancer Res. 2017, 15, 78–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.-P.; Goh, B.-C. Exosome-Mediated Metastasis: From Epithelial–Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Fridman, W.-H.; Pagès, F. The Adaptive Immunologic Microenvironment in Colorectal Cancer: A Novel Perspective. Cancer Res. 2007, 67, 1883–1886. [Google Scholar] [CrossRef] [Green Version]
- Gest, C.; Mirshahi, P.; Li, H.; Pritchard, L.-L.; Joimel, U.; Blot, E.; Chidiac, J.; Poletto, B.; Vannier, J.-P.; Varin, R.; et al. Ovarian cancer: Stat3, RhoA and IGF-IR as therapeutic targets. Cancer Lett. 2012, 317, 207–217. [Google Scholar] [CrossRef]
- Ye, F.; Chen, H.-Z.; Xie, X.; Ye, D.-F.; Lu, W.-G.; Ding, Z.-M. Vascular endothelial growth factor (VEGF) and ovarian carcinoma cell supernatant activate signal transducers and activators of transcription (STATs) via VEGF receptor-2 (KDR) in human hemopoietic progenitor cells. Gynecol. Oncol. 2004, 94, e125. [Google Scholar] [CrossRef]
- Tang, S.; Xiang, T.; Huang, S.; Zhou, J.; Wang, Z.; Xie, R.; Long, H.; Zhu, B. Ovarian cancer stem-like cells differentiate into endothelial cells and participate in tumor angiogenesis through autocrine CCL5 signaling. Cancer Lett. 2016, 376, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Langley, R.R.; Fidler, I.J. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005, 65, 10794–10800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Fu, S. Role of cancer-associated fibroblasts in tumor structure, composition and the microenvironment in ovarian cancer. Oncol. Lett. 2019, 18, 2173–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, F.; Cui, J.-Y.; Chen, L.; Chen, Y.-T.; Liu, B.-W. CAFs enhance paclitaxel resistance by inducing EMT through the IL-6/JAK2/STAT3 pathway. Oncol. Rep. 2018, 39, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Guo, B.-Y.; Zhang, S. Cancer-associated fibroblasts attenuate Cisplatin-induced apoptosis in ovarian cancer cells by promoting STAT3 signaling. Biochem. Biophys. Res. Commun. 2016, 470, 947–954. [Google Scholar] [CrossRef]
- Zhou, B.; Sun, C.; Li, N.; Shan, W.; Lu, H.; Guo, L.; Guo, E.; Xia, M.; Weng, D.; Meng, L.; et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int. J. Oncol. 2016, 48, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladányi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Kim, H.S.; Kim, S.; Haegeman, G.; Tsang, B.K.; Dhanasekaran, D.N.; Song, Y.S. Adipose Stromal Cells from Visceral and Subcutaneous Fat Facilitate Migration of Ovarian Cancer Cells via IL-6/JAK2/STAT3 Pathway. Cancer Res. Treat. 2017, 49, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, Y.; Odunsi, K.; Chen, W.; Peng, G.; Matsuzaki, J.; Wang, R.-F. Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 15505–15510. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 Regulates Cytokine-mediated Generation of Inflammatory Helper T Cells. J. Boil. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.-R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting Edge: An In Vivo Requirement for STAT3 Signaling in TH17 Development and TH17-Dependent Autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Huang, C.; Wang, X.-J.; Xin, D.E.; Wang, L.-S.; Zou, Q.C.; Zhang, Y.-N.S.; Tan, M.-D.; Wang, Y.-M.; Zhao, T.C.; et al. Lysyl Oxidase 3 Is a Dual-Specificity Enzyme Involved in STAT3 Deacetylation and Deacetylimination Modulation. Mol. Cell 2017, 65, 296–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25− Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-β Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Dercamp, C.; Chemin, K.; Caux, C.; Trinchieri, G.; Vicari, A.P. Distinct and Overlapping Roles of Interleukin-10 and CD25+ Regulatory T Cells in the Inhibition of Antitumor CD8 T-Cell Responses. Cancer Res. 2005, 65, 8479–8486. [Google Scholar] [CrossRef] [Green Version]
- Kinjyo, I.; Inoue, H.; Hamano, S.; Fukuyama, S.; Yoshimura, T.; Koga, K.; Takaki, H.; Himeno, K.; Takaesu, G.; Kobayashi, T.; et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor–β1. J. Exp. Med. 2006, 203, 1021–1031. [Google Scholar] [CrossRef]
- Kosack, L.; Wingelhofer, B.; Popa, A.; Orlova, A.; Agerer, B.; Vilagos, B.; Majek, P.; Parapatics, K.; Lercher, A.; Ringler, A.; et al. The ERBB-STAT3 Axis Drives Tasmanian Devil Facial Tumor Disease. Cancer Cell 2019, 35, 125–139.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, H.; Kato, T.; Tawara, I.; Ikeda, H.; Kuribayashi, K.; Allen, P.M.; Schreiber, R.D.; Old, L.J.; Shiku, H. IFN-γ Controls the Generation/Activation of CD4+CD25+ Regulatory T Cells in Antitumor Immune Response. J. Immunol. 2005, 175, 4433–4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xie, X.; Lu, W.-G.; Ye, D.-F.; Chen, H.-Z.; Li, X.; Cheng, Q. Ovarian carcinoma cells inhibit T cell proliferation: Suppression of IL-2 receptor beta and gamma expression and their JAK-STAT signaling pathway. Life Sci. 2004, 74, 1739–1749. [Google Scholar] [CrossRef]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The Ratios of CD8+ T Cells to CD4+CD25+ FOXP3+ and FOXP3- T Cells Correlate with Poor Clinical Outcome in Human Serous Ovarian Cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Cui, Y.; Li, X.; Cao, X.; Chen, A.; Xu, C.; Cao, J.; Luo, X. Co-culture of ovarian cancer stem-like cells with macrophages induced SKOV3 cells stemness via IL-8/STAT3 signaling. Biomed. Pharmacother. 2018, 103, 262–271. [Google Scholar] [CrossRef]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2012, 12, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Tweardy, D.J. STAT3 Inhibitors in Cancer: A Comprehensive Update; Humana Press: Cham, Switzerland, 2016; pp. 95–161. ISBN 978-3-319-42949-6. [Google Scholar]
- Wong, A.L.; Hirpara, J.L.; Pervaiz, S.; Eu, J.-Q.; Sethi, G.; Goh, B.-C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coward, J.; Kulbe, H.; Chakravarty, P.; Leader, D.; Vassileva, V.; Leinster, D.A.; Thompson, R.; Schioppa, T.; Nemeth, J.; Vermeulen, J.; et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 2011, 17, 6083–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, E.S.; Wen, W.; Dellinger, T.H.; Wu, J.; Lu, S.A.; Jove, R.; Yim, J.H. Ruxolitinib synergistically enhances the anti-tumor activity of paclitaxel in human ovarian cancer. Oncotarget 2018, 9, 24304–24319. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Shahda, S.; Beck, T.; Uppal, N.; Cohen, S.J.; Donehower, R.; Gabayan, A.; Assad, A.; Switzky, J.; Zhen, H.; et al. A Phase Ib/II Study of the JAK1 Inhibitor, Itacitinib, plus nab-Paclitaxel and Gemcitabine in Advanced Solid Tumors. Oncologist 2019, 24, 14-e10. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Luwor, R.; Burns, C.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 2018, 9, 16599–16618. [Google Scholar] [CrossRef]
- Liu, L.; Gaboriaud, N.; Vougogianopoulou, K.; Tian, Y.; Wu, J.; Wen, W.; Skaltsounis, L.; Jove, R. MLS-2384, a new 6-bromoindirubin derivative with dual JAK/Src kinase inhibitory activity, suppresses growth of diverse cancer cells. Cancer Biol. Ther. 2014, 15, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; Liang, W.; Wu, J.; Kowolik, C.M.; Buettner, R.; Scuto, A.; Hsieh, M.-Y.; Hong, H.; Brown, C.E.; Forman, S.J.; et al. Targeting JAK1/STAT3 signaling suppresses tumor progression and metastasis in a peritoneal model of human ovarian cancer. Mol. Cancer Ther. 2014, 13, 3037–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plimack, E.R.; Lorusso, P.M.; McCoon, P.; Tang, W.; Krebs, A.D.; Curt, G.; Eckhardt, S.G. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. Oncologist 2013, 18, 819–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedemans, R.; Dijkgraaf, E.M.; Santegoets, S.J.A.M.; Reyners, A.K.L.; Wouters, M.C.A.; Kenter, G.G.; Van Erkel, A.R.; Van Poelgeest, M.I.E.; Nijman, H.W.; Van Der Hoeven, J.J.M.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elnaggar, A.C.; Saini, U.; Naidu, S.; Wanner, R.; Sudhakar, M.; Fowler, J.; Nagane, M.; Kuppusamy, P.; Cohn, D.E.; Selvendiran, K. Anticancer potential of diarylidenyl piperidone derivatives, HO-4200 and H-4318, in cisplatin resistant primary ovarian cancer. Cancer Boil. Ther. 2016, 17, 1107–1115. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Ahmed, S.; Dayton, A.; Kuppusamy, M.L.; Rivera, B.K.; Kálai, T.; Hideg, K.; Kuppusamy, P. HO-3867, a curcumin analog, sensitizes cisplatin-resistant ovarian carcinoma, leading to therapeutic synergy through STAT3 inhibition. Cancer Boil. Ther. 2011, 12, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Selvendiran, K.; Tong, L.; Bratasz, A.; Kuppusamy, M.L.; Ahmed, S.; Ravi, Y.; Trigg, N.J.; Rivera, B.K.; Kálai, T.; Hideg, K.; et al. Anticancer efficacy of a difluorodiarylidenyl piperidone (HO-3867) in human ovarian cancer cells and tumor xenografts. Mol. Cancer Ther. 2010, 9, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- McCann, G.A.; Naidu, S.; Rath, K.S.; Bid, H.K.; Tierney, B.J.; Suarez, A.; Varadharaj, S.; Zhang, J.; Hideg, K.; Houghton, P.; et al. Targeting constitutively-activated STAT3 in hypoxic ovarian cancer, using a novel STAT3 inhibitor. Oncoscience 2014, 1, 216–228. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wang, F.; Wen, Z.; Shi, M.; Zhang, H. Blockage of STAT3 Signaling Pathway with a Decoy Oligodeoxynucleotide Inhibits Growth of Human Ovarian Cancer Cells. Cancer Investig. 2013, 32, 8–12. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, X.; Xu, X.; Shen, L.; Yao, Y.; Yang, Z.; Liu, P. STAT3 Decoy Oligodeoxynucleotides-Loaded Solid Lipid Nanoparticles Induce Cell Death and Inhibit Invasion in Ovarian Cancer Cells. PLoS ONE 2015, 10, e0124924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, P.; Zhang, B.; Wang, A.; Yang, M. Role of STAT3 decoy oligodeoxynucleotides on cell invasion and chemosensitivity in human epithelial ovarian cancer cells. Cancer Genet. Cytogenet. 2010, 197, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-H.; Xiang, Y.; Yu, C.-X.; Li, J.-P.; Li, H.; Nie, Q.; Hu, P.; Zhou, J.; Zhang, T.-C. STAT3 is required for MiR-17-5p-mediated sensitization to chemotherapy-induced apoptosis in breast cancer cells. Oncotarget 2014, 5, e15763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Bi, X.; Gao, G.; Sun, L. miRNA-133b and miRNA-135a induce apoptosis via the JAK2/STAT3 signaling pathway in human renal carcinoma cells. Biomed. Pharmacother. 2016, 84, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, J.; Mueller, A.C.; Dey, B.; Yang, Y.; Lee, D.-H.; Hachmann, J.; Finderle, S.; Park, D.M.; Christensen, J.; et al. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014, 21, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.-Y.; Yang, L.; Liu, X.-J.; Wang, X.-Z.; Pan, Y.-X.; Luo, J.-M. The Long Noncoding RNA MEG3 and its Target miR-147 Regulate JAK/STAT Pathway in Advanced Chronic Myeloid Leukemia. EBioMedicine 2018, 34, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Gheysarzadeh, A.; Yazdanparast, R. STAT5 Reactivation by Catechin Modulates H2O2-Induced Apoptosis Through miR-182/FOXO1 Pathway in SK-N-MC Cells. Cell Biochem. Biophys. 2015, 71, 649–656. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, W.; Zhang, G.; Xu, S.; Gao, Q.; Yang, Z. MicroRNA-204 targets JAK2 in breast cancer and induces cell apoptosis through the STAT3/BCl-2/survivin pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 5017–5025. [Google Scholar]
- Zhao, B.; Dong, A.-S. MiR-874 inhibits cell growth and induces apoptosis by targeting STAT3 in human colorectal cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 269–277. [Google Scholar]
- Zaanan, A.; Okamoto, K.; Kawakami, H.; Khazaie, K.; Huang, S.; Sinicrope, F.A. The Mutant KRAS Gene Up-regulates BCL-XL Protein via STAT3 to Confer Apoptosis Resistance That Is Reversed by BIM Protein Induction and BCL-XL Antagonism*. J. Boil. Chem. 2015, 290, 23838–23849. [Google Scholar] [CrossRef] [Green Version]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Nuñez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Ray, R.M.; Johnson, L.R. STAT3-mediated transcription of Bcl-2, Mcl-1 and c-IAP2 prevents apoptosis in polyamine-depleted cells. Biochem. J. 2005, 392, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanuti, P.; Bertagnolo, V.; Pierdomenico, L.; Bascelli, A.; Santavenere, E.; Alinari, L.; Capitani, S.; Miscia, S.; Marchisio, M. Enhancement of TRAIL cytotoxicity by AG-490 in human ALL cells is characterized by downregulation of cIAP-1 and cIAP-2 through inhibition of Jak2/Stat3. Cell Res. 2009, 19, 1079–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, T.; Iwasaki, T.; Okada, T.; Hashimoto, T.; Moon, Y.; Sakaguchi, M.; Fukami, Y.; Nishigori, C.; Oka, M. High expression of Mcl-1L via the MEK-ERK-phospho-STAT3 (Ser727) pathway protects melanocytes and melanoma from UVB-induced apoptosis. Genes Cells 2016, 21, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, T.; Boyd, S.; Mijatov, B.; Gowrishankar, K.; Snoyman, S.; Pupo, G.; Scolyer, R.; Mann, G.; Kefford, R.; Zhang, X.; et al. Mutant B-RAF-Mcl-1 survival signaling depends on the STAT3 transcription factor. Oncogene 2014, 33, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, S.; Terui, K.; Zhang, H.Q.; Enosawa, S.; Ogawa, W.; Inoue, H.; Okuyama, T.; Takeda, K.; Akira, S.; Ogino, T.; et al. Stat3 protects against Fas-induced liver injury by redox-dependent and -independent mechanisms. J. Clin. Investig. 2003, 112, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Zou, M.; Xu, C.; Li, H.; Zhang, X.; Fan, W. 3,3′-Diindolylmethane suppresses ovarian cancer cell viability and metastasis and enhances chemotherapy sensitivity via STAT3 and Akt signaling in vitro and in vivo. Arch. Biochem. Biophys. 2018, 651. [Google Scholar] [CrossRef]
- Ganji, P.N.; Park, W.; Wen, J.; Mahaseth, H.; Landry, J.; Farris, A.B.; Willingham, F.; Sullivan, P.S.; Proia, D.A.; El-Hariry, I.; et al. Antiangiogenic effects of ganetespib in colorectal cancer mediated through inhibition of HIF-1α and STAT-3. Angiogenesis 2013, 16, 903–917. [Google Scholar] [CrossRef]
- Wincewicz, A.; Koda, M.; Sulkowska, M.; Kanczuga-Koda, L.; Sulkowski, S. Comparison of STAT3 with HIF-1alpha, Ob and ObR expressions in human endometrioid adenocarcinomas. Tissue Cell 2008, 40, 405–410. [Google Scholar] [CrossRef]
- Xie, T.-X.; Huang, F.-J.; Aldape, K.D.; Kang, S.-H.; Liu, M.; Gershenwald, J.E.; Xie, K.; Sawaya, R.; Huang, S. Activation of Stat3 in Human Melanoma Promotes Brain Metastasis. Cancer Res. 2006, 66, 3188–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.-X.; Wei, D.; Liu, M.; Gao, A.C.; Ali-Osman, F.; Sawaya, R.; Huang, S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004, 23, 3550–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, A.; Wang, S.C.; Morris, J.P.; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Guo, F.; Xu, G.; Ma, J.; Shao, F. STAT3 cooperates with Twist to mediate epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Oncol. Rep. 2015, 33, 1872–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Wang, Q.; Wang, B.; Sun, Q.; He, Z.; Hong, J.; Kuehn, F.; Liu, E.; Zhang, Z. IGF-1 induces the epithelial-mesenchymal transition via Stat5 in hepatocellular carcinoma. Oncotarget 2014, 5, 111922–111930. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Erreni, M.; Kamal, M.A.; Porta, C.; Marchesi, F.; Pesce, S.; Pasqualini, F.; Schiarea, S.; Chiabrando, C.; Mantovani, A.; et al. Differential role of Interleukin-1 and Interleukin-6 in K-Ras-driven pancreatic carcinoma undergoing mesenchymal transition. OncoImmunology 2017, 7, e1388485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talati, P.G.; Gu, L.; Ellsworth, E.M.; Girondo, M.A.; Trerotola, M.; Hoang, D.T.; Leiby, B.; Dagvadorj, A.; McCue, P.A.; Lallas, C.D.; et al. Jak2-Stat5a/b Signaling Induces Epithelial-to-Mesenchymal Transition and Stem-Like Cell Properties in Prostate Cancer. Am. J. Pathol. 2015, 185, 2505–2522. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.S.; Brim, H.; Sherif, Z.A. Co-overexpression of Janus kinase 2 and signal transducer and activator of transcription 5a promotes differentiation of mammary cancer cells through reversal of epithelial–mesenchymal transition. Cancer Sci. 2008, 99, 272–279. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 Feed-Forward Loop Drives Tumorigenesis and Metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Hadjidaniel, M.D.; Muthugounder, S.; Hung, L.T.; Sheard, M.A.; Shirinbak, S.; Chan, R.Y.; Nakata, R.; Borriello, L.; Malvar, J.; Kennedy, R.J.; et al. Tumor-associated macrophages promote neuroblastoma via STAT3 phosphorylation and up-regulation of c-MYC. Oncotarget 2014, 5, 91516–91529. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2014, 5, e20741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Aksoy, I.; Gonnot, F.; Osteil, P.; Aubry, M.; Hamela, C.; Rognard, C.; Hochard, A.; Voisin, S.; Fontaine, E.; et al. Reinforcement of STAT3 activity reprogrammes human embryonic stem cells to naive-like pluripotency. Nat. Commun. 2015, 6, e7095. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Xiang, T.; Zhao, Z.; Lin, K.; Yin, P.; Jiang, L.; Liang, Z.; Zhu, B. Autocrine interleukin-23 promotes self-renewal of CD133+ ovarian cancer stem-like cells. Oncotarget 2016, 7, e76006. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, H.; Ding, J.; Nie, S.; Wang, L.; Zhang, L.; Ren, S. Celastrol strongly inhibits proliferation, migration and cancer stem cell properties through suppression of Pin1 in ovarian cancer cells. Eur. J. Pharmacol. 2019, 842, 146–156. [Google Scholar] [CrossRef]

| Category | Drug | Target | Phase | Histology | Remark | References |
|---|---|---|---|---|---|---|
| Upstream ligand | Siltuximab (CNTO 328) | IL-6 | Phase II | 19 serous, 1 CC |
| [146] |
| Tociclizumab | IL-6R | Phase I | 15 serous, 3 CC, 3 EM |
| [154] | |
| Upstream kinase inhibitors | Ruxolitinib (INC424) | JAK 1, JAK 2 | Phase I/II | Not specified |
| [147] NCT02713386. |
| Tofacitinib (CP690550) | JAK3>JAK1>>JAK2 | Not tested in EOC | - |
| ||
| Baricitinib (INCB28050) | JAK 1, JAK 2 | Not tested in EOC | - |
| ||
| Itacitinib (ABT494) | JAK 1, JAK 2 | Not tested in EOC | - |
| NCT02646748 | |
| Momelotinib (CYT387) | JAK 1, JAK 2 | Preclinical | Serous and CC cell lines |
| [150,155] | |
| MLS2384 | c-SRC, JAK 2, TYK2 | Preclinical | Not specified |
| [151] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-J.; Sundararajan, V.; Sheu, B.-C.; Huang, R.Y.-J.; Wei, L.-H. Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity. Cancers 2020, 12, 24. https://doi.org/10.3390/cancers12010024
Wu C-J, Sundararajan V, Sheu B-C, Huang RY-J, Wei L-H. Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity. Cancers. 2020; 12(1):24. https://doi.org/10.3390/cancers12010024
Chicago/Turabian StyleWu, Chin-Jui, Vignesh Sundararajan, Bor-Ching Sheu, Ruby Yun-Ju Huang, and Lin-Hung Wei. 2020. "Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity" Cancers 12, no. 1: 24. https://doi.org/10.3390/cancers12010024