Gene Expression Alterations during Development of Castration-Resistant Prostate Cancer Are Detected in Circulating Tumor Cells


Abstract
:1. Introduction
2. Results
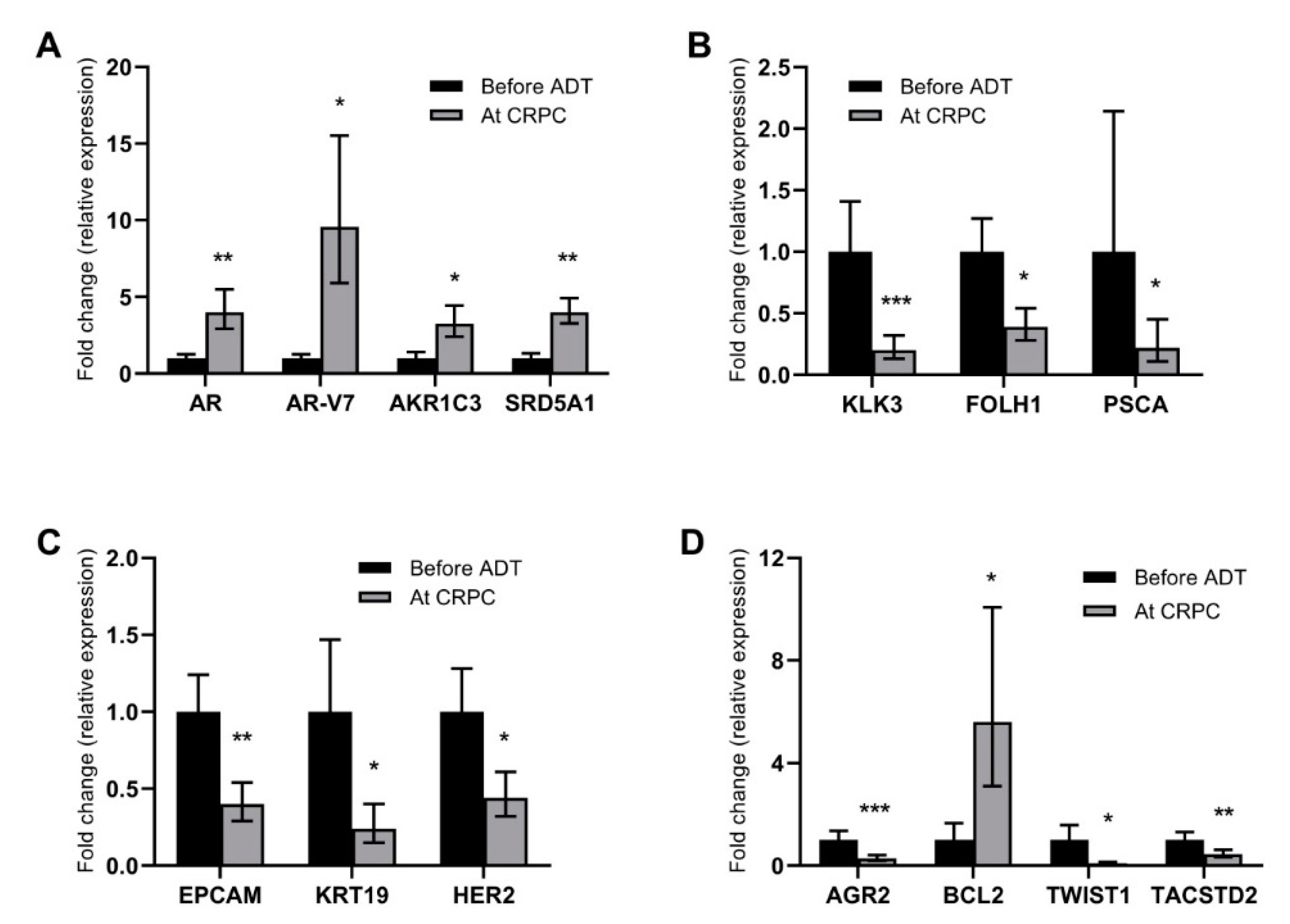
2.1. Alterations in Gene Expression during ADT
2.2. Gene Expression in CTCs as Prognostic Markers for Survival
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. CTC Isolation and cDNA Synthesis
4.3. Gene Expression Profiling
4.4. Definition of CTC Load as Basis for Normalization and Handling of Missing Signals
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, N.D.; Spears, M.R.; Clarke, N.W.; Dearnaley, D.P.; De Bono, J.S.; Gale, J.; Hetherington, J.; Hoskin, P.J.; Jones, R.J.; Laing, R.; et al. Survival with newly diagnosed metastatic prostate cancer in the “docetaxel era”: Data from 917 patients in the control arm of the stampede trial (mrc pr08, cruk/06/019). Eur. Urol. 2015, 67, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [Green Version]
- Titus, M.A.; Schell, M.J.; Lih, F.B.; Tomer, K.B.; Mohler, J.L. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4653–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, C.W.; Johnson, R.T., Jr.; Mohler, J.L.; French, F.S.; Wilson, E.M. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001, 61, 2892–2898. [Google Scholar] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Linja, M.J.; Savinainen, K.J.; Saramaki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef]
- Barrie, S.E.; Potter, G.A.; Goddard, P.M.; Haynes, B.P.; Dowsett, M.; Jarman, M. Pharmacology of novel steroidal inhibitors of cytochrome p450(17) alpha (17 alpha-hydroxylase/c17-20 lyase). J. Steroid Biochem. Mol. Biol. 1994, 50, 267–273. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. Ar-v7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of ar-v7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Ritch, C.R.; Cookson, M.S. Advances in the management of castration resistant prostate cancer. BMJ 2016, 355, i4405. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, A.; Larsson, K.; Mansson, M.; Bjorkman, J.; Rohlova, E.; Ahs, D.; Brisby, H.; Damber, J.E.; Welen, K. Circulating tumor cells mirror bone metastatic phenotype in prostate cancer. Oncotarget 2018, 9, 29403–29413. [Google Scholar] [CrossRef]
- Bu, H.; Bormann, S.; Schafer, G.; Horninger, W.; Massoner, P.; Neeb, A.; Lakshmanan, V.K.; Maddalo, D.; Nestl, A.; Sultmann, H.; et al. The anterior gradient 2 (agr2) gene is overexpressed in prostate cancer and may be useful as a urine sediment marker for prostate cancer detection. Prostate 2011, 71, 575–587. [Google Scholar] [CrossRef]
- Chanda, D.; Lee, J.H.; Sawant, A.; Hensel, J.A.; Isayeva, T.; Reilly, S.D.; Siegal, G.P.; Smith, C.; Grizzle, W.; Singh, R.; et al. Anterior gradient protein-2 is a regulator of cellular adhesion in prostate cancer. PLoS ONE 2014, 9, e89940. [Google Scholar] [CrossRef] [Green Version]
- Kani, K.; Malihi, P.D.; Jiang, Y.; Wang, H.; Wang, Y.; Ruderman, D.L.; Agus, D.B.; Mallick, P.; Gross, M.E. Anterior gradient 2 (agr2): Blood-based biomarker elevated in metastatic prostate cancer associated with the neuroendocrine phenotype. Prostate 2013, 73, 306–315. [Google Scholar] [CrossRef]
- De Bono, J.S.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.V.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef] [Green Version]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [Green Version]
- Jernberg, E.; Thysell, E.; Bovinder Ylitalo, E.; Rudolfsson, S.; Crnalic, S.; Widmark, A.; Bergh, A.; Wikstrom, P. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS ONE 2013, 8, e77407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Titus, M.A.; Gregory, C.W.; Ford, O.H., 3rd; Schell, M.J.; Maygarden, S.J.; Mohler, J.L. Steroid 5alpha-reductase isozymes i and ii in recurrent prostate cancer. Clin. Cancer Res. 2005, 11, 4365–4371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofland, J.; van Weerden, W.M.; Dits, N.F.; Steenbergen, J.; van Leenders, G.J.; Jenster, G.; Schroder, F.H.; de Jong, F.H. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010, 70, 1256–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsiades, N.; Sung, C.C.; Schultz, N.; Danila, D.C.; He, B.; Eedunuri, V.K.; Fleisher, M.; Sander, C.; Sawyers, C.L.; Scher, H.I. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012, 72, 6142–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallee, M.P.; Visser-de Jong, E.; van der Korput, J.A.; van der Kwast, T.H.; ten Kate, F.J.; Schroeder, F.H.; Trapman, J. Variation of prostate-specific antigen expression in different tumour growth patterns present in prostatectomy specimens. Urol. Res. 1990, 18, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stege, R.H.; Tribukait, B.; Carlstrom, K.A.; Grande, M.; Pousette, A.H. Tissue psa from fine-needle biopsies of prostatic carcinoma as related to serum psa, clinical stage, cytological grade, and DNA ploidy. Prostate 1999, 38, 183–188. [Google Scholar] [CrossRef]
- Chen, C.L.; Mahalingam, D.; Osmulski, P.; Jadhav, R.R.; Wang, C.M.; Leach, R.J.; Chang, T.C.; Weitman, S.D.; Kumar, A.P.; Sun, L.; et al. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of emt-related genes in metastatic prostate cancer. Prostate 2013, 73, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Markou, A.; Lazaridou, M.; Paraskevopoulos, P.; Chen, S.; Swierczewska, M.; Budna, J.; Kuske, A.; Gorges, T.M.; Joosse, S.A.; Kroneis, T.; et al. Multiplex gene expression profiling of in vivo isolated circulating tumor cells in high-risk prostate cancer patients. Clin. Chem. 2018, 64, 297–306. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: Implications for androgen-deprivation therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of n-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific ar45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Goodman, O.B., Jr.; Symanowski, J.T.; Loudyi, A.; Fink, L.M.; Ward, D.C.; Vogelzang, N.J. Circulating tumor cells as a predictive biomarker in patients with hormone-sensitive prostate cancer. Clin. Genitourin. Cancer 2011, 9, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Nordin, A.; Wang, W.; Welen, K.; Damber, J.E. Midkine is associated with neuroendocrine differentiation in castration-resistant prostate cancer. Prostate 2013, 73, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Gong, A.; Cheville, J.C.; Smith, D.I.; Young, C.Y. Agr2, an androgen-inducible secretory protein overexpressed in prostate cancer. Genes Chromosomes Cancer 2005, 43, 249–259. [Google Scholar] [CrossRef]
- Gravdal, K.; Halvorsen, O.J.; Haukaas, S.A.; Akslen, L.A. A switch from e-cadherin to n-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin. Cancer Res. 2007, 13, 7003–7011. [Google Scholar] [CrossRef] [Green Version]
- Jennbacken, K.; Tesan, T.; Wang, W.; Gustavsson, H.; Damber, J.E.; Welen, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Linder, A.; Hagberg Thulin, M.; Damber, J.E.; Welen, K. Analysis of regulator of g-protein signalling 2 (rgs2) expression and function during prostate cancer progression. Sci. Rep. 2018, 8, 17259. [Google Scholar] [CrossRef] [Green Version]
- Cottard, F.; Asmane, I.; Erdmann, E.; Bergerat, J.P.; Kurtz, J.E.; Ceraline, J. Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PLoS ONE 2013, 8, e63466. [Google Scholar] [CrossRef]
- Cottard, F.; Madi-Berthelemy, P.O.; Erdmann, E.; Schaff-Wendling, F.; Keime, C.; Ye, T.; Kurtz, J.E.; Ceraline, J. Dual effects of constitutively active androgen receptor and full-length androgen receptor for n-cadherin regulation in prostate cancer. Oncotarget 2017, 8, 72008–72020. [Google Scholar] [CrossRef] [Green Version]
- Josefsson, A.; Damber, J.E.; Welen, K. Ar-v7 expression in circulating tumor cells as a potential prognostic marker in metastatic hormone-sensitive prostate cancer. Acta Oncol. 2019, 58, 1660–1664. [Google Scholar] [CrossRef]
- Josefsson, A.; Linder, A.; Flondell-Site, D.; Canesin, G.; Stiehm, A.; Anand, A.; Bjartell, A.; Damber, J.E.; Welen, K. Circulating tumor cells as a marker for progression-free survival in metastatic castration-naïve prostate cancer. Prostate 2017, 77, 849–858. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Clinical Variables | Number or Median (Q1; Q3) |
|---|---|
| PSA before androgen deprivation therapy (ADT) | 590 ng/mL (270; 1900) |
| Gleason sum | |
| 6–7 | 4 |
| 8 | 9 |
| 9–10 | 17 |
| x | 7 |
| Type of ADT | |
| GnRH agonist | 28 |
| GnRH antagonist | 7 |
| Orchidectomy | 2 |
| Time from ADT to CRPC relapse | 8.4 months (4.9; 12.9) |
| Castration-resistant prostate cancer CRPC Therapy | |
| Total androgen blockade (bicalutamide) | 25 |
| Abiraterone acetate or enzalutamide | 11 |
| Radium-223 | 9 |
| Docetaxel or cabazitaxel | 8 |
| Cyclophosphamide | 2 |
| None | 5 |
| Time from ADT to prostate cancer death or last follow-up (cancer-specific survival, CSS) (n = 35 *) | 25.7 months (13.6; 39.3) |
| Total follow-up time (n = 37) | 24.1 months (11.1; 39.0) |
| Gene | Before ADT (n = 40) (A) | At CRPC Relapse (n = 32) | Pairs Included in Comparison (A vs. CRPC) ** | ||
|---|---|---|---|---|---|
| Detected Signals | Substituted * Missing Signals | Detected Signals | Substituted * Missing Signals | ||
| AGR2 | 30 | 8 | 14 | 18 | 25 |
| AHR | 13 | 24 | 9 | 20 | 13 |
| AKR1C3 | 23 | 17 | 21 | 11 | 24 |
| AR | 23 | 18 | 17 | 14 | 19 |
| ARV7 | 9 | 26 | 10 | 8 | 12 |
| AURKA | 21 | 19 | 19 | 10 | 22 |
| BCL2 | 16 | 23 | 17 | 11 | 21 |
| CDH1 | 22 | 14 | 14 | 15 | 17 |
| CDH2 | 4 | 30 | 4 | 24 | 4 |
| DDR1 | 13 | 23 | 13 | 16 | 14 |
| EGFR | 12 | 9 | 8 | 9 | 9 |
| EMP2 | 22 | 16 | 13 | 17 | 16 |
| EPCAM | 36 | 2 | 23 | 9 | 29 |
| FOLH1 | 30 | 7 | 15 | 14 | 22 |
| UPA | 11 | 25 | 10 | 15 | 13 |
| HER2 | 24 | 16 | 16 | 16 | 21 |
| KLK3 | 33 | 5 | 21 | 11 | 27 |
| KRT19 | 28 | 9 | 17 | 14 | 25 |
| MDK | 29 | 11 | 16 | 15 | 25 |
| POU5F1 | 19 | 20 | 18 | 14 | 22 |
| PSCA | 17 | 20 | 9 | 21 | 14 |
| SNAI1 | 10 | 19 | 4 | 22 | 9 |
| SPINK1 | 20 | 20 | 12 | 20 | 18 |
| SRD5A1 | 25 | 15 | 27 | 4 | 28 |
| TACSTD2 | 29 | 9 | 18 | 8 | 24 |
| TOP2A | 26 | 13 | 18 | 8 | 21 |
| TP53 | 34 | 6 | 28 | 4 | 28 |
| TUBB3 | 21 | 15 | 14 | 16 | 21 |
| TWIST1 | 21 | 17 | 11 | 21 | 19 |
| VEGFA | 24 | 16 | 17 | 15 | 23 |
| Parameter | Time to CRPC | Cancer-Specific Survival | ||
|---|---|---|---|---|
| p-Value | Hazard Ratio | p-Value | Hazard Ratio | |
| (95% Confidence Interval) | (95% Confidence Interval) | |||
| Average Cq | 0.0005 | 1.22 (1.09–1.38) | 0.0003 | 1.24 (1.10–1.39) |
| EPCAM | 0.012 | 1.32 (1.06–1.65) | 0.002 | 1.52 (1.16–1.97) |
| MDK | 0.061 | 1.11 (0.99–1.25) | 0.008 | 1.25 (1.06–1.47) |
| TWIST | 0.537 | 0.96 (0.85–1.09) | 0.050 | 0.87 (0.75–1.00) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Josefsson, A.; Larsson, K.; Freyhult, E.; Damber, J.-E.; Welén, K. Gene Expression Alterations during Development of Castration-Resistant Prostate Cancer Are Detected in Circulating Tumor Cells. Cancers 2020, 12, 39. https://doi.org/10.3390/cancers12010039
Josefsson A, Larsson K, Freyhult E, Damber J-E, Welén K. Gene Expression Alterations during Development of Castration-Resistant Prostate Cancer Are Detected in Circulating Tumor Cells. Cancers. 2020; 12(1):39. https://doi.org/10.3390/cancers12010039
Chicago/Turabian StyleJosefsson, Andreas, Karin Larsson, Eva Freyhult, Jan-Erik Damber, and Karin Welén. 2020. "Gene Expression Alterations during Development of Castration-Resistant Prostate Cancer Are Detected in Circulating Tumor Cells" Cancers 12, no. 1: 39. https://doi.org/10.3390/cancers12010039
APA StyleJosefsson, A., Larsson, K., Freyhult, E., Damber, J.-E., & Welén, K. (2020). Gene Expression Alterations during Development of Castration-Resistant Prostate Cancer Are Detected in Circulating Tumor Cells. Cancers, 12(1), 39. https://doi.org/10.3390/cancers12010039