Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. ROS1 Is Present in Mitochondria
2.2. ROS1 Contains a Novel Mitochondria-Targeting Signal
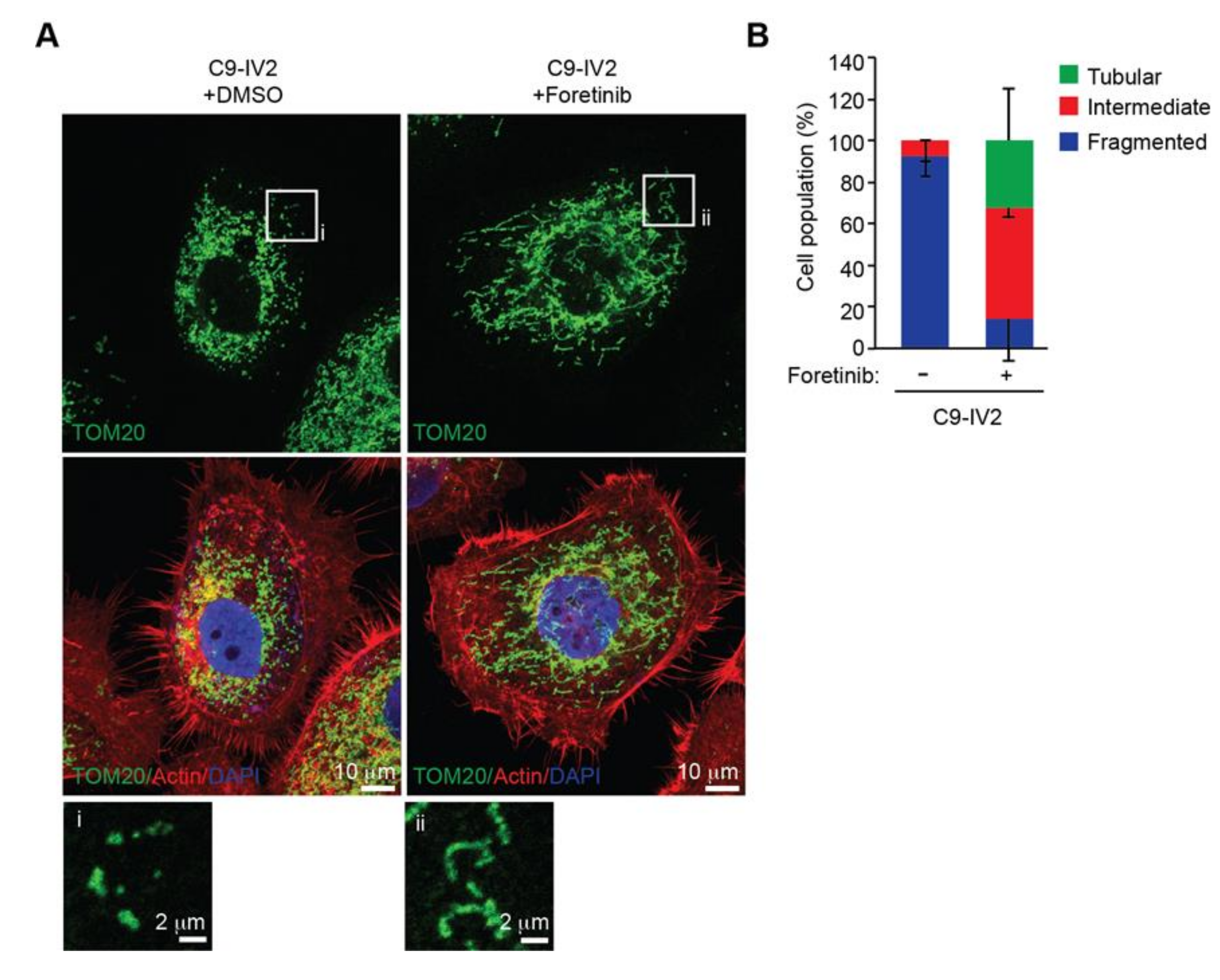
2.3. Mitochondria are More Fragmented in Highly Invasive Oral Cancer Cells
2.4. ROS1 Expression Induces Mitochondrial Morphogenesis in OSCC Cells
2.5. ROS1 Enhances Mitochondrial Bioenergetics and Metabolic Plasticity but Reduces Mitochondrial Biogenesis in OSCC Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Plasmids
4.4. Knockdown of ROS1
4.5. Reverse Transcription and Semiquantitative Real-time PCR (Q-PCR)
4.6. Immunoblotting
4.7. Immunofluorescence Staining, Confocal Microscopy, Mitochondrial Morphology, and Colocalization Analysis
4.8. Subcellular Fractionation and Proteinase K Treatment of Mitochondria
4.9. Measurement of Oxygen Consumption Rate (OCR)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Godeny, M. Prognostic factors in advanced pharyngeal and oral cavity cancer; significance of multimodality imaging in terms of 7th edition of TNM. Cancer Imaging 2014, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeffler-Ragg, J.; Schwentner, I.; Sprinzl, G.M.; Zwierzina, H. EGFR inhibition as a therapy for head and neck squamous cell carcinoma. Expert Opin. Investig. Drugs 2008, 17, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar] [CrossRef]
- Geva, Y.; Schuldiner, M. The back and forth of cargo exit from the endoplasmic reticulum. Curr. Biol. CB 2014, 24, R130–R136. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.H.; Chang, Y.J.; Huang, W.C.; Jang, T.H.; Kung, H.J.; Wang, W.C.; Yang, M.H.; Lin, M.C.; Huang, S.F.; Chou, S.W.; et al. EZH2-mediated upregulation of ROS1 oncogene promotes oral cancer metastasis. Oncogene 2017, 36, 6542–6554. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Chan, S.H.; Jang, T.H.; Chang, J.W.; Ko, Y.C.; Yen, T.C.; Chiang, S.L.; Chiang, W.F.; Shieh, T.Y.; Liao, C.T.; et al. miRNA-491-5p and GIT1 serve as modulators and biomarkers for oral squamous cell carcinoma invasion and metastasis. Cancer Res. 2014, 74, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.D.; Doebele, R.C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef] [Green Version]
- Acquaviva, J.; Wong, R.; Charest, A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim. Biophys. Acta 2009, 1795, 37–52. [Google Scholar] [CrossRef]
- Charest, A.; Kheifets, V.; Park, J.; Lane, K.; McMahon, K.; Nutt, C.L.; Housman, D. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc. Natl. Acad. Sci. USA 2003, 100, 916–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neel, D.S.; Allegakoen, D.V.; Olivas, V.; Mayekar, M.K.; Hemmati, G.; Chatterjee, N.; Blakely, C.M.; McCoach, C.E.; Rotow, J.K.; Le, A.; et al. Differential Subcellular Localization Regulates Oncogenic Signaling by ROS1 Kinase Fusion Proteins. Cancer Res. 2019, 79, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Sun, Y.; Wang, L.Z.; Yu, Y.C.; Ding, X. Cytoplasmic c-ros oncogene 1 receptor tyrosine kinase expression may be associated with the development of human oral squamous cell carcinoma. Oncol. Lett. 2015, 10, 934–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, D.H.; Hwang, I. Specific targeting of proteins to outer envelope membranes of endosymbiotic organelles, chloroplasts, and mitochondria. Front. Plant Sci. 2014, 5, 173. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Ma, J.T.; Zhang, X.Y.; Cao, R.; Sun, L.; Jing, W.; Zhao, J.Z.; Zhang, S.L.; Huang, L.T.; Han, C.B. Effects of Dynamin-related Protein 1 Regulated Mitochondrial Dynamic Changes on Invasion and Metastasis of Lung Cancer Cells. J. Cancer 2019, 10, 4045–4053. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Cannino, G.; Ciscato, F.; Masgras, I.; Sanchez-Martin, C.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333. [Google Scholar] [CrossRef]
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930. [Google Scholar] [CrossRef] [Green Version]
- Lehuede, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, H.; Cai, Y.; Ye, J.T.; Liu, Z.P.; Lu, J.; Huang, X.Y.; Feng, X.J.; Gao, H.; Chen, S.R.; et al. Mitochondrial binding of alpha-enolase stabilizes mitochondrial membrane: Its role in doxorubicin-induced cardiomyocyte apoptosis. Arch. Biochem. Biophys. 2014, 542, 46–55. [Google Scholar] [CrossRef]
- Kim, Y.M.; Youn, S.W.; Sudhahar, V.; Das, A.; Chandhri, R.; Cuervo Grajal, H.; Kweon, J.; Leanhart, S.; He, L.; Toth, P.T.; et al. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018, 23, 3565–3578. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Hung, M.C. Nuclear functions and subcellular trafficking mechanisms of the epidermal growth factor receptor family. Cell Biosci. 2012, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Chem. 2009, 284, 36592–36604. [Google Scholar] [CrossRef] [Green Version]
- Che, T.F.; Lin, C.W.; Wu, Y.Y.; Chen, Y.J.; Han, C.L.; Chang, Y.L.; Wu, C.T.; Hsiao, T.H.; Hong, T.M.; Yang, P.C. Mitochondrial translocation of EGFR regulates mitochondria dynamics and promotes metastasis in NSCLC. Oncotarget 2015, 6, 37349–37366. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.K.; Hung, M.C. Proteolytic cleavage, trafficking, and functions of nuclear receptor tyrosine kinases. FEBS J. 2015, 282, 3693–3721. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pages, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Liu, C.J.; Chiu, C.P.; Chang, S.M.; Lu, S.Y.; Chen, Y.J. Establishment of OC3 oral carcinoma cell line and identification of NF-kappa B activation responses to areca nut extract. J. Oral Pathol. Med. 2004, 33, 79–86. [Google Scholar] [CrossRef]
- Lu, Y.C.; Chen, Y.J.; Wang, H.M.; Tsai, C.Y.; Chen, W.H.; Huang, Y.C.; Fan, K.H.; Tsai, C.N.; Huang, S.F.; Kang, C.J.; et al. Oncogenic function and early detection potential of miRNA-10b in oral cancer as identified by microRNA profiling. Cancer Prev. Res. 2012, 5, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, L.; Herrington, J.; Carter-Su, C. SH2-B is required for nerve growth factor-induced neuronal differentiation. J. Biol. Chem. 1999, 274, 10590–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-J.; Chen, K.-W.; Chen, L. Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma. Cancers 2020, 12, 2845. https://doi.org/10.3390/cancers12102845
Chang Y-J, Chen K-W, Chen L. Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma. Cancers. 2020; 12(10):2845. https://doi.org/10.3390/cancers12102845
Chicago/Turabian StyleChang, Yu-Jung, Kuan-Wei Chen, and Linyi Chen. 2020. "Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma" Cancers 12, no. 10: 2845. https://doi.org/10.3390/cancers12102845