Olaparib Synergizes the Anticancer Activity of Daunorubicin via Interaction with AKR1C3



Abstract
:Sample summary
Abstract

1. Introduction
2. Results
2.1. Screening for Interactions of Anthracycline Reductases with Olaparib
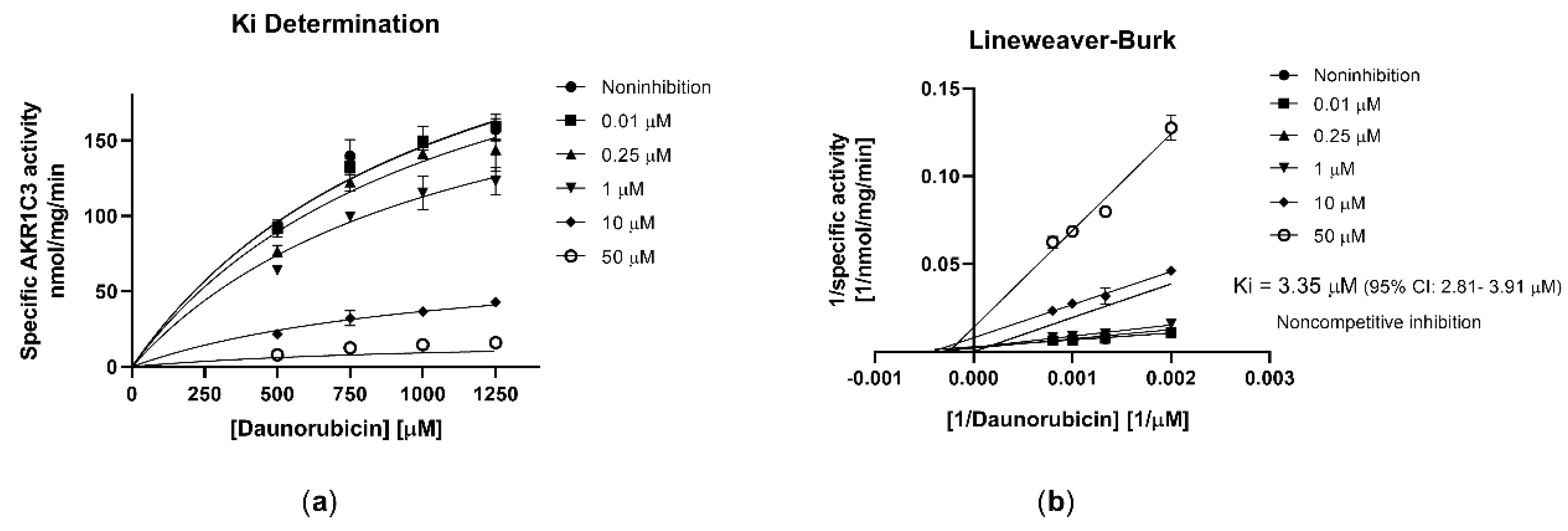
2.2. Olaparib Potently Inhibits Human Recombinant AKR1C3
2.3. Effect of Olaparib on AKR1C3-Mediated Daunorubicin Metabolism in HCT116 Cells
2.4. Olaparib Synergize with Daunorubicin due to Interaction with AKR1C3
2.5. Assessment of AKR1C3 Expression following Exposure to Olaparib
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Cultures
4.3. Cloning, Overexpression, and Purification of Recombinant CREs
4.4. Incubation Assay with Recombinant CREs
4.5. Docking Studies
4.6. Transient Transfection of HCT116 Cell Line
4.7. AKR1C3 Inhibitory Assay in HCT116 Cells
4.8. XTT Proliferation Assay
4.9. Drug Combination
4.10. Induction Studies
4.11. Ultra-High-Performance Liquid Chromatography (UHPLC)
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Piska, K.; Koczurkiewicz, P.; Bucki, A.; Wójcik-Pszczoła, K.; Kołaczkowski, M.; Pękala, E. Metabolic Carbonyl Reduction of Anthracyclines—Role in Cardiotoxicity and Cancer Resistance. Reducing Enzymes as Putative Targets for Novel Cardioprotective and Chemosensitizing Agents. Investig. New Drugs 2017, 35, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Funes, H.; Coronado, C. Role of Anthracyclines in the Era of Targeted Therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J.; Doles, J.; Zender, L.; Xue, W.; Ma, B.; McCombie, W.R.; Hannon, G.J.; Lowe, S.W.; Hemann, M.T. Topoisomerase Levels Determine Chemotherapy Response In Vitro and In Vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 9053–9058. [Google Scholar] [CrossRef] [Green Version]
- Capelôa, T.; Benyahia, Z.; Zampieri, L.X.; Blackman, M.C.; Sonveaux, P. Metabolic and Non-Metabolic Pathways That Control Cancer Resistance to Anthracyclines. Semin. Cell Dev. Biol. 2020, 98, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, C.M.; White, C.C.; Merrill, G.F.; Kavanagh, T.J. Metabolism of Doxorubicin to the Cardiotoxic Metabolite Doxorubicinol Is Increased in a Mouse Model of Chronic Glutathione Deficiency: A Potential Role for Carbonyl Reductase 3. Chem. Biol. Interact. 2014, 234, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malátková, P.; Wsól, V. Carbonyl Reduction Pathways in Drug Metabolism. Drug Metab. Rev. 2013, 46, 96–123. [Google Scholar] [CrossRef]
- Edwardson, D.W.; Narendrula, R.; Chewchuk, S.; Mispel-Beyer, K.; Mapletoft, J.P.; Parissenti, A.M. Role of Drug Metabolism in the Cytotoxicity and Clinical Efficacy of Anthracyclines. Curr. Drug Metab. 2015, 16, 412–426. [Google Scholar] [CrossRef] [Green Version]
- Bains, O.S.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Naturally Occurring Variants of Human Aldo-Keto Reductases with Reduced In Vitro Metabolism of Daunorubicin and Doxorubicin. J. Pharmacol. Exp. Ther. 2010, 335, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Hofman, J.; Malcekova, B.; Skarka, A.; Novotna, E.; Wsol, V. Anthracycline Resistance Mediated by Reductive Metabolism in Cancer Cells: The Role of Aldo-Keto Reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–248. [Google Scholar] [CrossRef]
- Desmond, J.C.; Mountford, J.C.; Drayson, M.T.; Walker, E.A.; Hewison, M.; Ride, J.P.; Luong, Q.T.; Hayden, R.E.; Vanin, E.F.; Bunce, C.M. The Aldo-Keto Reductase AKR1C3 is a Novel Suppressor of Cell Differentiation That Provides a Plausible Target for the Non-Cyclooxygenase-Dependent Antineoplastic Actions of Nonsteroidal Anti-Inflammatory Drugs. Cancer Res. 2003, 63, 505–512. [Google Scholar] [PubMed]
- Birtwistle, J.; Hayden, R.E.; Khanim, F.L.; Green, R.M.; Pearce, C.; Davies, N.J.; Wake, N.; Schrewe, H.; Ride, J.P.; Chipman, J.K.; et al. The Aldo-Keto Reductase AKR1C3 Contributes to 7,12-Dimethylbenz(a)Anthracene-3,4-Dihydrodiol Mediated Oxidative DNA Damage in Myeloid Cells: Implications for Leukemogenesis. Mutat. Res. Mol. Mech. Mutagen. 2009, 662, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Byrns, M.C. Steroid Hormone Transforming Aldo-Keto Reductases and Cancer. Ann. N.Y. Acad. Sci. 2009, 1155, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-Q.; Gu, X.; Gao, X.-S.; Li, Y.; Yu, H.; Xiong, W.; Yu, H.; Wang, W.; Li, Y.; Teng, Y.; et al. Overexpression of AKR1C3 Significantly Enhances Human Prostate Cancer Cells Resistance to Radiation. Oncotarget 2016, 7, 48050–48058. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Wada, Y.; Endo, S.; Soda, M.; El-Kabbani, O.; Hara, A. Aldo–Keto Reductase 1B10 and Its Role in Proliferation Capacity of Drug-Resistant Cancers. Front. Pharmacol. 2012, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Bortolozzi, R.; Bresolin, S.; Rampazzo, E.; Paganin, M.; Maule, F.; Mariotto, E.; Boso, D.; Minuzzo, S.; Agnusdei, V.; Viola, G.; et al. AKR1C Enzymes Sustain Therapy Resistance in Paediatric T-ALL. Br. J. Cancer 2018, 118, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Varatharajan, S.; Abraham, A.; Zhang, W.; Shaji, R.V.; Ahmed, R.; Abraham, A.; George, B.; Srivastava, A.; Chandy, M.; Mathews, V.; et al. Carbonyl Reductase 1 Expression Influences Daunorubicin Metabolism in Acute Myeloid Leukemia. Eur. J. Clin. Pharmacol. 2012, 68, 1577–1586. [Google Scholar] [CrossRef]
- Marin, J.J.; Briz, O.; Rodríguez-Macias, G.; Díez-Martín, J.L.; Macias, R. Role of Drug Transport and Metabolism in the Chemoresistance of Acute Myeloid Leukemia. Blood Rev. 2016, 30, 55–64. [Google Scholar] [CrossRef]
- Verma, K.; Zang, T.; Penning, T.M.; Trippier, P.C. Potent and Highly Selective Aldo–Keto Reductase 1C3 (AKR1C3) Inhibitors Act as Chemotherapeutic Potentiators in Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. J. Med. Chem. 2019, 62, 3590–3616. [Google Scholar] [CrossRef]
- Penning, T.M. The Aldo-Keto Reductases (AKRs): Overview. Chem. Interact. 2015, 234, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Drew, Y. The Development of PARP Inhibitors in Ovarian Cancer: From Bench to Bedside. Br. J. Cancer 2015, 113, S3–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; Van Der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; De Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Bixel, K.; Hays, J.L. Olaparib in the Management of Ovarian Cancer. Pharm. Pers. Med. 2015, 8, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.N.J.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallotta, V.; Conte, C.; D’Indinosante, M.; Capoluongo, E.; Minucci, A.; De Rose, A.M.; Ardito, F.; Giuliante, F.; Di Giorgio, A.; Zannoni, G.F.; et al. Prognostic Factors Value of Germline and Somatic Brca in Patients Undergoing Surgery for Recurrent Ovarian Cancer with Liver Metastases. Eur. J. Surg. Oncol. 2019, 45, 2096–2102. [Google Scholar] [CrossRef]
- Faraoni, I.; Consalvo, M.I.; Aloisio, F.; Fabiani, E.; Giansanti, M.; Di Cristino, F.; Falconi, G.; Tentori, L.; Veroli, A.; Curzi, P.; et al. Cytotoxicity and Differentiating Effect of the Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Myelodysplastic Syndromes. Cancers 2019, 11, 1373. [Google Scholar] [CrossRef] [Green Version]
- Maifrede, S.; Nieborowska-Skorska, M.; Sullivan-Reed, K.; Dasgupta, Y.; Podszywalow-Bartnicka, P.; Le, B.V.; Solecka, M.; Lian, Z.; Belyaeva, E.A.; Nersesyan, A.; et al. Tyrosine Kinase Inhibitor–Induced Defects in DNA Repair Sensitize FLT3(ITD)-Positive Leukemia Cells to PARP1 Inhibitors. Blood 2018, 132, 67–77. [Google Scholar] [CrossRef]
- Faraoni, I.; Aloisio, F.; De Gabrieli, A.; Consalvo, M.I.; Lavorgna, S.; Voso, M.T.; Albano, F.; Graziani, G. The Poly(ADP-Ribose) Polymerase Inhibitor Olaparib Induces up-Regulation of Death Receptors in Primary Acute Myeloid Leukemia Blasts by NF-κB Activation. Cancer Lett. 2018, 423, 127–138. [Google Scholar] [CrossRef]
- Meng, X.W.; Koh, B.D.; Zhang, J.-S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-Ribose) Polymerase Inhibitors Sensitize Cancer Cells to Death Receptor-mediated Apoptosis by Enhancing Death Receptor Expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Bae, J.S.; Kim, K.M.; Moon, Y.J.; Park, S.-H.; Ha, S.H.; Hussein, U.K.; Zhang, Z.; Park, H.S.; Park, B.-H.; et al. The PARP Inhibitor Olaparib Potentiates the Effect of the DNA Damaging Agent Doxorubicin in Osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 107. [Google Scholar] [CrossRef]
- Eetezadi, S.; Evans, J.C.; Shen, Y.T.; De Souza, R.; Piquette-Miller, M.; Allen, C. Ratio-Dependent Synergism of a Doxorubicin and Olaparib Combination in 2D and Spheroid Models of Ovarian Cancer. Mol. Pharm. 2018, 15, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Del Conte, G.; Sessa, C.; Von Moos, R.; Viganò, L.; Digena, T.; Locatelli, A.; Gallerani, E.; Fasolo, A.; Tessari, A.; Cathomas, R.; et al. Phase I Study of Olaparib in Combination With Liposomal Doxorubicin in Patients With Advanced Solid Tumours. Br. J. Cancer 2014, 111, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassner, N.; Huse, K.; Martin, H.-J.; Gödtel-Armbrust, U.; Metzger, A.; Meineke, I.; Brockmöller, J.; Klein, K.; Zanger, U.M.; Maser, E.; et al. Carbonyl Reductase 1 Is a Predominant Doxorubicin Reductase in the Human Liver. Drug Metab. Dispos. 2008, 36, 2113–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrns, M.C.; Jin, Y.; Penning, T.M. Inhibitors of Type 5 17β-Hydroxysteroid Dehydrogenase (AKR1C3): Overview and Structural Insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovering, A.L.; Ride, J.P.; Bunce, C.M.; Desmond, J.C.; Cummings, S.M.; White, S.A. Crystal Structures of Prostaglandin D211-Ketoreductase (AKR1C3) in Complex with the Nonsteroidal Anti-Inflammatory Drugs Flufenamic Acid and Indomethacin. Cancer Res. 2004, 64, 1802–1810. [Google Scholar] [CrossRef] [Green Version]
- Krogh-Madsen, M.; Bender, B.C.; Jensen, M.K.; Nielsen, O.J.; Friberg, L.E.; Honoré, P.H. Population Pharmacokinetics of Cytarabine, Etoposide, and Daunorubicin in the Treatment for Acute Myeloid Leukemia. Cancer Chemother. Pharmacol. 2012, 69, 1155–1163. [Google Scholar] [CrossRef]
- Food and Drug Administration. Approval Date(s) and History, Letters, Labels, Reviews for NDA 208558.Efficacy-New Indication. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=208558 (accessed on 15 January 2020).
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, C.C.; Moore, K. Olaparib: An Oral PARP-1 and PARP-2 Inhibitor with Promising Activity in Ovarian Cancer. Future Oncol. 2015, 11, 747–757. [Google Scholar] [CrossRef]
- Hintzpeter, J.; Seliger, J.M.; Hofman, J.; Martin, H.-J.; Wsol, V.; Maser, E. Inhibition of Human Anthracycline Reductases by Emodin—A Possible Remedy for Anthracycline Resistance. Toxicol. Appl. Pharmacol. 2016, 293, 21–29. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.; De Bono, J. A Decade of Clinical Development of PARP Inhibitors in Perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Aschenbrenner, D.S. Olaparib Approved for Metastatic Pancreatic Cancer. AJN Am. J. Nurs. 2020, 120, 22–23. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. CPMP/EWP/560/95/Rev. 1 Corr. 2**. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.p (accessed on 20 May 2020).
- Adeniji, A.O.; Chen, M.; Penning, T.M. AKR1C3 as a Target in Castrate Resistant Prostate Cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. Aldo-Keto Reductase (AKR) 1C3 Inhibitors: A Patent Review. Expert Opin. Ther. Pat. 2017, 27, 1329–1340. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Yu, E.Y. AR-Signaling in Human Malignancies: Prostate Cancer and Beyond. Cancers 2017, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Novotna, R.; Wsol, V.; Xiong, G.; Maser, E. Inactivation of the Anticancer Drugs Doxorubicin and Oracin by Aldo–Keto Reductase (AKR) 1C3. Toxicol. Lett. 2008, 181, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Škarydová, L.; Nobilis, M.; Wsól, V. Role of Carbonyl Reducing Enzymes in the Phase I Biotransformation of the Non-Steroidal Anti-Inflammatory Drug Nabumetone In Vitro. Xenobiotica 2012, 43, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking With a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer, v20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Novotná, E.; Büküm, N.; Hofman, J.; Flaxová, M.; Kouklíková, E.; Louvarová, D.; Wsol, V. Aldo-Keto Reductase 1C3 (AKR1C3): A Missing Piece of the Puzzle in the Dinaciclib Interaction Profile. Arch. Toxicol. 2018, 92, 2845–2857. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Inhibition % Olaparib (10 µM) | Inhibition % Olaparib (50 µM) |
|---|---|---|
| AKR1C3 | 76.0 ± 0.32 | 91.4 ± 0.97 |
| AKR1B10 | 0.0 | 7.7 ± 0.33 |
| AKR1A1 | 1.8 ± 0.14 | 18.2 ± 0.39 |
| AKR1B1 | 0.0 | 8.1 ± 0.83 |
| CBR1 | 0.0 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavares, T.S.; Hofman, J.; Lekešová, A.; Želazková, J.; Wsól, V. Olaparib Synergizes the Anticancer Activity of Daunorubicin via Interaction with AKR1C3. Cancers 2020, 12, 3127. https://doi.org/10.3390/cancers12113127
Tavares TS, Hofman J, Lekešová A, Želazková J, Wsól V. Olaparib Synergizes the Anticancer Activity of Daunorubicin via Interaction with AKR1C3. Cancers. 2020; 12(11):3127. https://doi.org/10.3390/cancers12113127
Chicago/Turabian StyleTavares, Tássia S., Jakub Hofman, Alžběta Lekešová, Jana Želazková, and Vladimír Wsól. 2020. "Olaparib Synergizes the Anticancer Activity of Daunorubicin via Interaction with AKR1C3" Cancers 12, no. 11: 3127. https://doi.org/10.3390/cancers12113127