In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead

 ,
,  and
and 
Abstract
:Simple Summary
Abstract
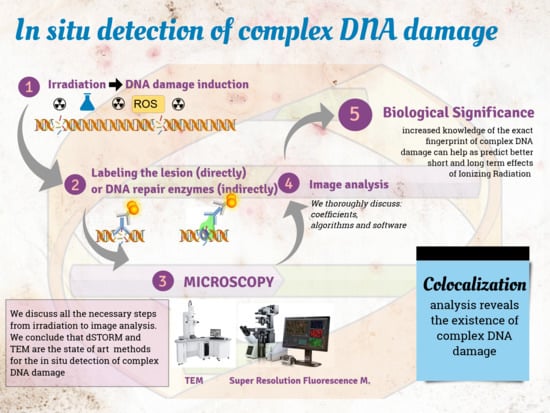
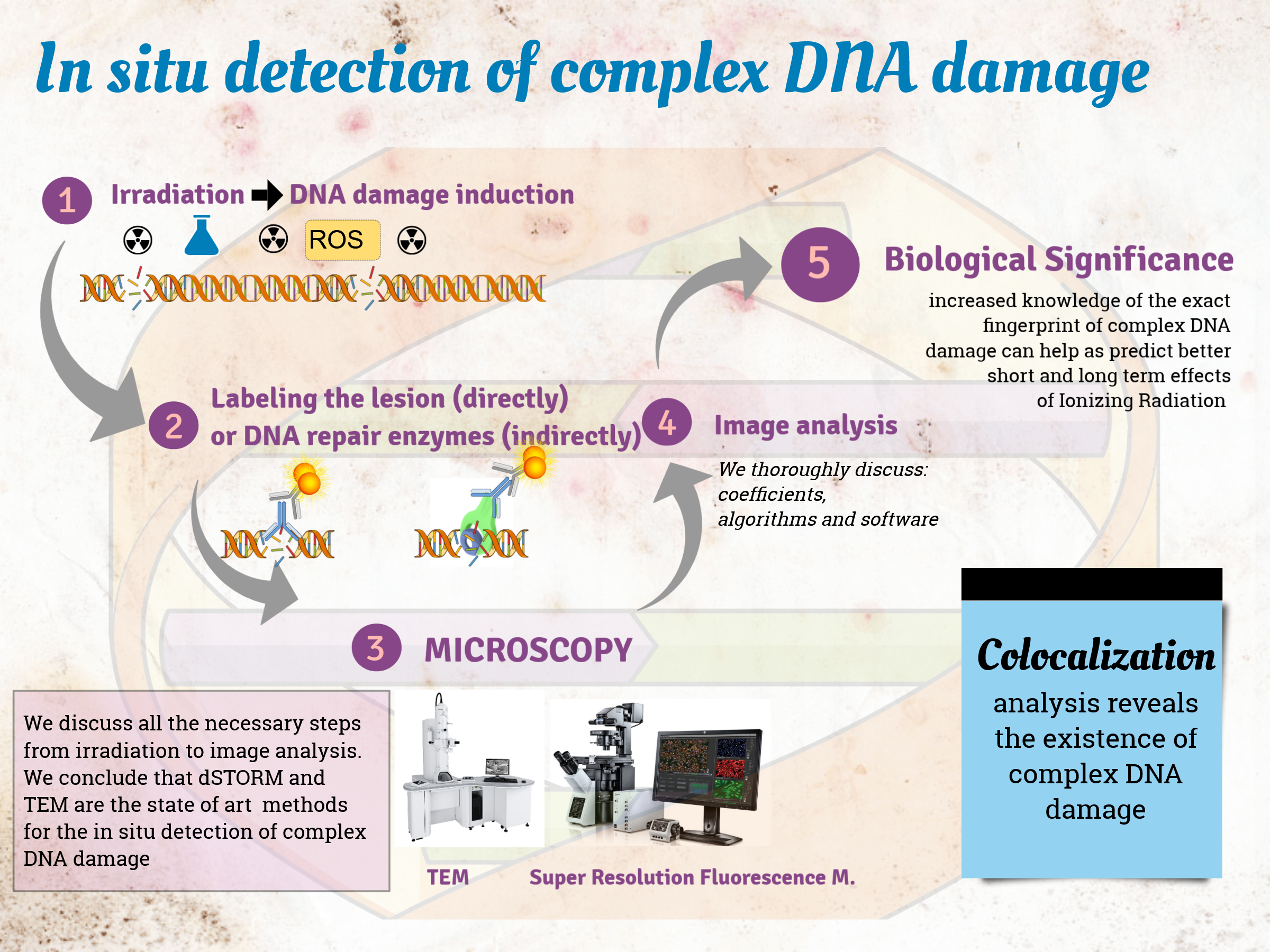
1. Introduction
1.1. General Concept for In Situ Detection of Complex DNA Damage
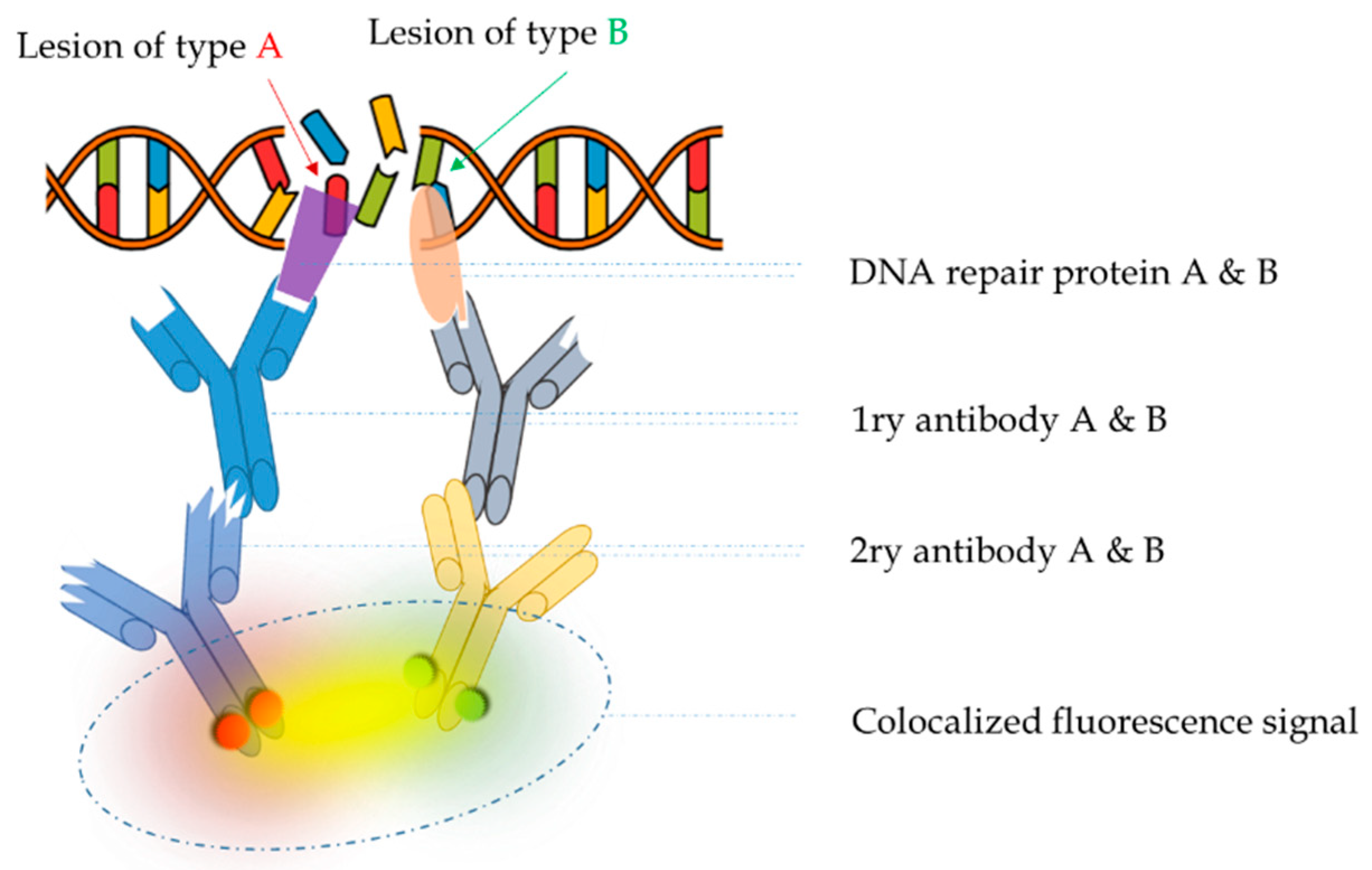
1.2. Colocalization: The Principle behind the In Situ Detection of Complex DNA Damage
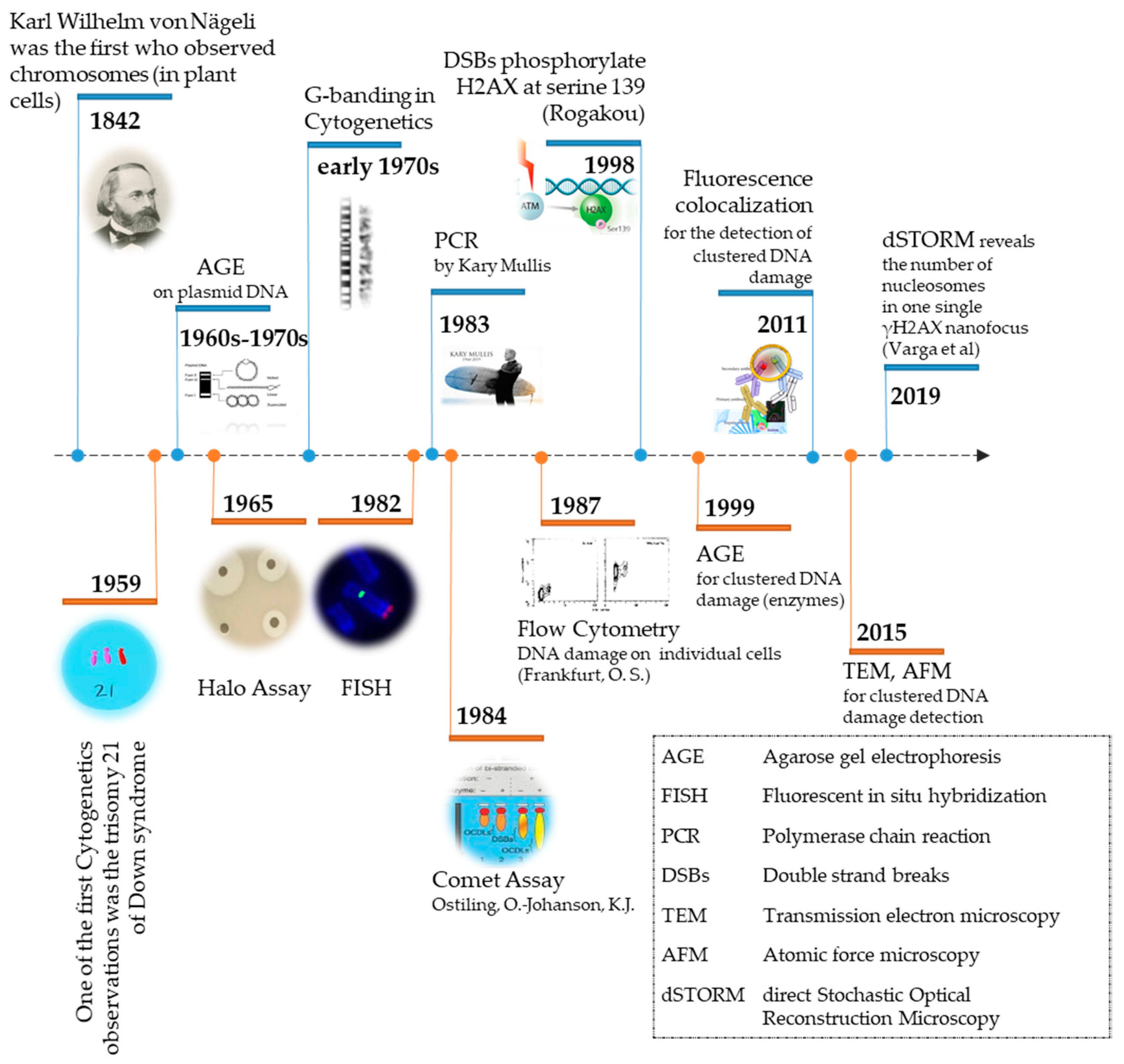
2. Damage Induction
3. Damage Visualization
3.1. Entity to Be Detected: Direct (the Damage Itself) or Indirect (via DNA Repair Proteins)
3.2. Labeling Techniques for Fixed and Live Cells
3.2.1. Immunolabeling of Fixed Cells
Fluorophore Conjugated Antibodies for In Situ Immunofluorescence
Nanoparticle Conjugated Antibodies for Transmission Electron Microscopy (TEM) Analysis
Proximity Ligation Assay
3.2.2. Live Cell Imaging
Encoding Fluorescence Labeled Proteins through Plasmid Transfection
Fluorogenic Dyes
Encoding Fluorescence Labeled Proteins Using CRISPR/Cas9
Fluorescence Recovery after Photobleaching (FRAP)
4. Imaging: Microscopy and Cameras
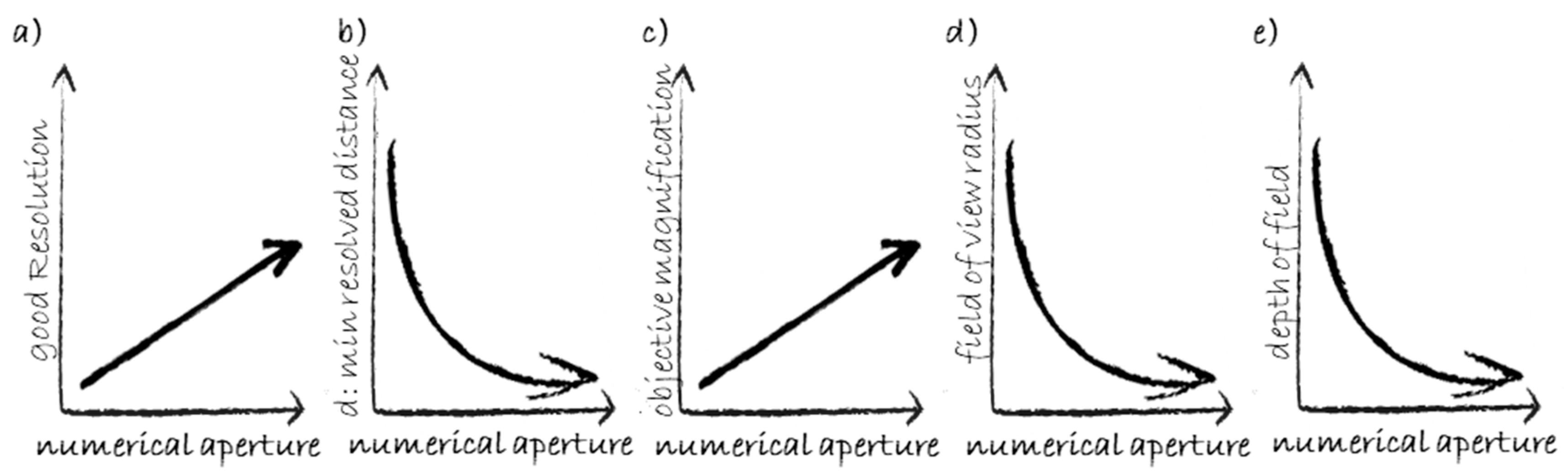
4.1. Conventional (Widefield) Fluorescence Microscopy and Basic Features
- Bright-field: When the specimen absorbs or scatters some photons and appear darker than its background, which appears bright; in the bright field image, the unscattered (transmitted) photons are selected to the detector and the scattered photons are blocked.
- In Dark-field mode the unscattered photons are excluded from the aperture and the scattered ones are selected instead. Hence, the areas around the sample do not scatter the light and they will appear dark, while the specimen will appear bright.
4.2. Confocal Microscopy
4.3. Super Resolution Microscopy (SRM)
4.3.1. Super Resolution: Beyond the Diffraction Limit
4.3.2. Far Field vs. Near Field and the Evanescence Illumination
4.3.3. Super Resolution: Stochastic Techniques for Single Molecule Detection
4.4. Non-Photon Microscopy
4.4.1. Scanning Probe
4.4.2. Electron Microscopy: TEM and SEM
Transmission Electron Microscopy-TEM
Scanning Electron Microscopy-SEM
4.5. Cameras and Photomultipliers
5. Image Analysis
5.1. Cell and Foci Recognition: Criteria, Algorithms, and Transformations
5.2. Colocalization Analysis: Coefficients, Parameters, and Methods
5.2.1. Threshold-Based Colocalization
- i.
- Pearson Correlation Coefficient:
- ii.
- (Manders) Overlap Coefficient, r:
- iii.
- (Manders) Overlap Coefficient, r2, k1, and k2:withand
- iv.
- (Manders) M1 and M2 overlap Coefficient:withand
- v.
- Costes’ Automatic Threshold
- vi.
- Van Steensel’s CCF
- vii.
- Cytofluorogram
- viii.
- Li’s ICQ
5.2.2. Topology-Based Colocalization
- i.
- Rcol
- ii.
- Pclc
- iii.
- Defining an Extra Type of Focus
5.3. Clustering Analysis
5.4. Co-Localization in Super Resolution Localization Microscopy
5.5. Software
5.5.1. ImageJ
5.5.2. CellProfiler
5.5.3. ColocalizR
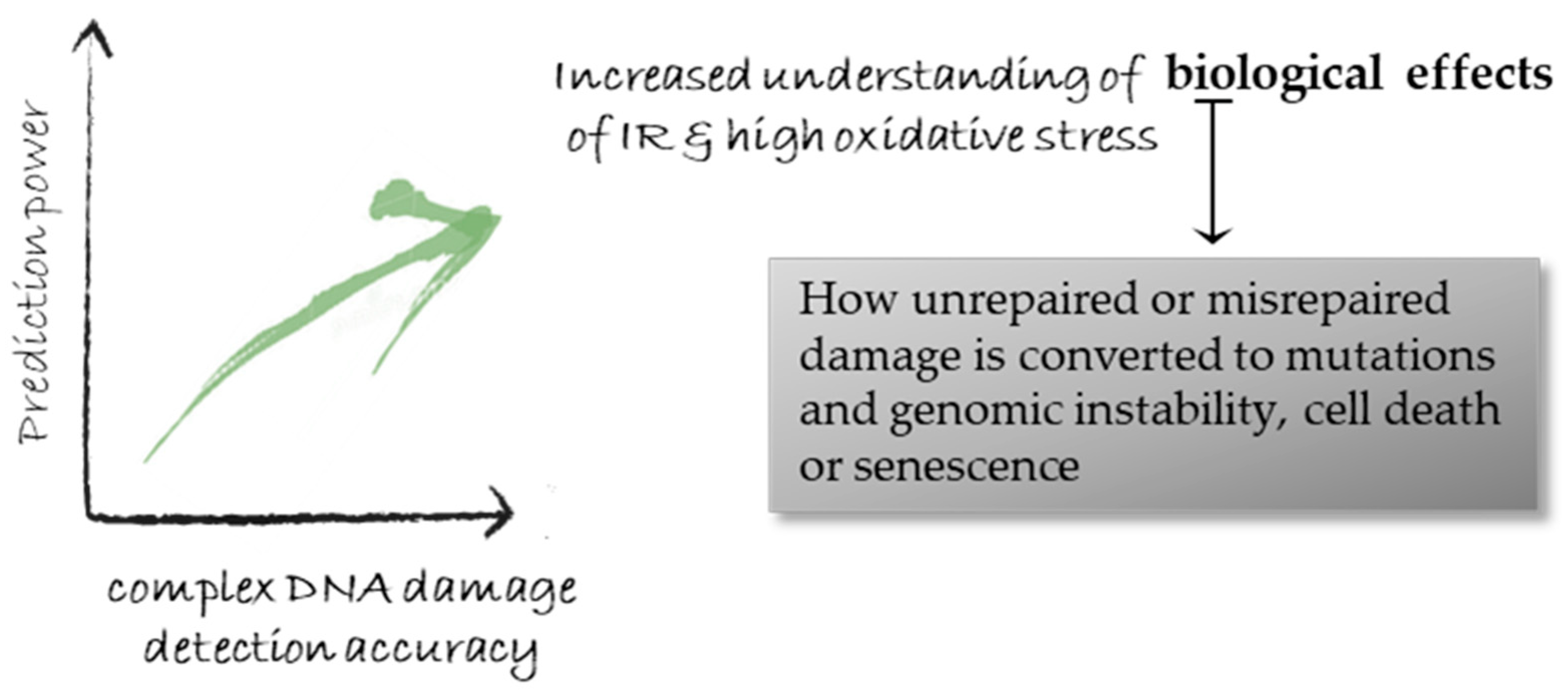
6. Biological and Clinical Importance of Progress in Signal Detection
7. Conclusions
7.1. Fixed Cells vs. Live Cell Imaging
7.2. Conventional Wide Field vs. Confocal Microscopy
7.3. Super Resolution-Wide Field-Stochastic-Single Molecule for In Situ DNA Damage Detection
7.4. The Persistent Weakness of Complex DNA Damage Detection
7.5. The Crucial Necessity to Study DNA Damage and Repair
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CCD | Charge-coupled Device |
| CMOS | Complementary Metal-oxide Semiconductor |
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-González, G.; Pérez-Plasencia, C. Strategies for the evaluation of DNA damage and repair mechanisms in cancer. Oncol. Lett. 2017, 13, 3982–3988. [Google Scholar] [CrossRef] [Green Version]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Kareva, I.G.; Naf, D.; Nowsheen, S.; Kryston, T.B.; Bonner, W.M.; Georgakilas, A.G.; Sedelnikova, O.A. Tumors induce complex DNA damage in distant proliferative tissues in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17992–17997. [Google Scholar] [CrossRef] [Green Version]
- Reddig, A.; Rübe, C.E.; Rödiger, S.; Schierack, P.; Reinhold, D.; Roggenbuck, D. DNA damage assessment and potential applications in laboratory diagnostics and precision medicine. J. Lab. Precis. Med. 2018, 3. [Google Scholar] [CrossRef]
- Aaij, C.; Borst, P. The gel electrophoresis of DNA. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1972, 269, 192–200. [Google Scholar] [CrossRef]
- Vinograd, J.; Lebowitz, J.; Radloff, R.; Watson, R.; Laipis, P. The twisted circular form of polyoma viral DNA. Proc. Natl. Acad. Sci. USA 1965, 53, 1104–1111. [Google Scholar] [CrossRef] [Green Version]
- Sestili, P. The fast-halo assay for the assessment of DNA damage at the single-cell level. Methods Mol. Biol. 2009, 521, 517–533. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA 2000, 97, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, I.; Nikitaki, Z.; Souli, M.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; O’Neill, P.; Stewart, R.D. Induction and repair of clustered DNA lesions: What do we know so far? Radiat. Res. 2013, 180, 100–109. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Bennett, P.V.; Sidorkina, O.; Laval, J. Clustered damages and total lesions induced in DNA by ionizing radiation: Oxidized bases and strand breaks. Biochemistry 2000, 39, 8026–8031. [Google Scholar] [CrossRef]
- Dong, Y.; Gao, Y.; Liu, W.; Gao, T.; Zheng, Y.; Sanche, L. Clustered DNA Damage Induced by 2-20 eV Electrons and Transient Anions: General Mechanism and Correlation to Cell Death. J. Phys. Chem. Lett. 2019, 10, 2985–2990. [Google Scholar] [CrossRef]
- Sutherland, B.M.; Georgakilas, A.G.; Bennett, P.V.; Laval, J.; Sutherland, J.C. Quantifying clustered DNA damage induction and repair by gel electrophoresis, electronic imaging and number average length analysis. Mutat. Res. 2003, 531, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Brabcová, K.P.; Sihver, L.; Ukraintsev, E.; Stepan, V.; Davidkova, M. Length computation of irradiated plasmid DNA molecules. Biointerphases 2018, 13, 061005. [Google Scholar] [CrossRef]
- Terzoudi, G.I.; Pantelias, G.; Darroudi, F.; Barszczewska, K.; Buraczewska, I.; Depuydt, J.; Georgieva, D.; Hadjidekova, V.; Hatzi, V.I.; Karachristou, I.; et al. Dose assessment intercomparisons within the RENEB network using G0-lymphocyte prematurely condensed chromosomes (PCC assay). Int. J. Radiat. Biol. 2017, 93, 48–57. [Google Scholar] [CrossRef] [Green Version]
- McNeil, N.; Ried, T. Novel molecular cytogenetic techniques for identifying complex chromosomal rearrangements: Technology and applications in molecular medicine. Expert Rev. Mol. Med. 2000, 2000, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar] [CrossRef] [Green Version]
- Ashley, T.; Plug, A.W.; Xu, J.; Solari, A.J.; Reddy, G.; Golub, E.I.; Ward, D.C. Dynamic changes in Rad51 distribution on chromatin during meiosis in male and female vertebrates. Chromosoma 1995, 104, 19–28. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Schultz, L.B.; Chehab, N.H.; Malikzay, A.; Halazonetis, T.D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000, 151, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Maser, R.S.; Monsen, K.J.; Nelms, B.E.; Petrini, J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997, 17, 6087–6096. [Google Scholar] [CrossRef] [Green Version]
- Nelms, B.E.; Maser, R.S.; MacKay, J.F.; Lagally, M.G.; Petrini, J.H. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 1998, 280, 590–592. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Georgescu, W.; Deschamps, T.; Yannone, S.M.; Costes, S.V. Mathematical Modeling for DNA Repair, Carcinogenesis and Cancer Detection. In Genomic Instability and Cancer Metastasis; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2015; pp. 75–93. [Google Scholar]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Mladenov, E.; Mangelis, A.; Laskaratou, D.A.; Fragkoulis, G.I.; Hellweg, C.E.; Martin, O.A.; Emfietzoglou, D.; et al. Measurement of complex DNA damage induction and repair in human cellular systems after exposure to ionizing radiations of varying linear energy transfer (LET). Free Radic. Res. 2016, 50, S64–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, G.A.; Siebenwirth, C.; Drexler, S.E.; Girst, S.; Greubel, C.; Dollinger, G.; Friedl, A.A. Live cell imaging at the Munich ion microbeam SNAKE—A status report. Radiat. Oncol. 2015, 10, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauptner, A.; Krucken, R.; Greubel, C.; Hable, V.; Dollinger, G.; Drexler, G.A.; Deutsch, M.; Lowe, R.; Friedl, A.A.; Dietzel, S.; et al. DNA-repair protein distribution along the tracks of energetic ions. Radiat. Prot. Dosim. 2006, 122, 147–149. [Google Scholar] [CrossRef]
- Friedland, W.; Kundrat, P.; Schmitt, E.; Becker, J.; Ilicic, K.; Greubel, C.; Reindl, J.; Siebenwirth, C.; Schmid, T.E.; Dollinger, G. Modeling Studies on Dicentrics Induction after Sub-Micrometer Focused Ion Beam Grid Irradiation. Radiat. Prot. Dosim. 2019, 183, 40–44. [Google Scholar] [CrossRef]
- Hong, Z.; Jiang, J.; Hashiguchi, K.; Hoshi, M.; Lan, L.; Yasui, A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J. Cell Sci. 2008, 121, 3146–3154. [Google Scholar] [CrossRef] [Green Version]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015, 11, 1486–1500. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrov, R.; Dotchev, A.; Poser, I.; Krastev, D.; Georgiev, G.; Panova, G.; Babukov, Y.; Danovski, G.; Dyankova, T.; Hubatsch, L.; et al. Protein Dynamics in Complex DNA Lesions. Mol. Cell 2018, 69, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, J.; Rudolph, J.; Jha, A.; Tay, J.W.; Dragavon, J.; Grumstrup, E.M.; Luger, K. Q-FADD: A Mechanistic Approach for Modeling the Accumulation of Proteins at Sites of DNA Damage. Biophys. J. 2019, 116, 2224–2233. [Google Scholar] [CrossRef]
- Jain, S.; Coulter, J.A.; Hounsell, A.R.; Butterworth, K.T.; McMahon, S.J.; Hyland, W.B.; Muir, M.F.; Dickson, G.R.; Prise, K.M.; Currell, F.J.; et al. Cell-specific radiosensitization by gold nanoparticles at megavoltage radiation energies. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef] [Green Version]
- Bellaiche, Y.; Mogila, V.; Perrimon, N. I-SceI endonuclease, a new tool for studying DNA double-strand break repair mechanisms in Drosophila. Genetics 1999, 152, 1037–1044. [Google Scholar]
- Honma, M.; Sakuraba, M.; Koizumi, T.; Takashima, Y.; Sakamoto, H.; Hayashi, M. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair 2007, 6, 781–788. [Google Scholar] [CrossRef]
- Iliakis, G.; Mladenova, V.; Sharif, M.; Chaudhary, S.; Mavragani, I.V.; Soni, A.; Saha, J.; Schipler, A.; Mladenov, E. Defined Biological Models of High-LET Radiation Lesions. Radiat. Prot. Dosim. 2018, 183, 60–68. [Google Scholar] [CrossRef]
- van den Berg, J.; G Manjón, A.; Kielbassa, K.; Feringa, F.M.; Freire, R.; Medema, R.H. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018, 46, 10132–10144. [Google Scholar] [CrossRef]
- Fenech, M.; Bonassi, S. The effect of age, gender, diet and lifestyle on DNA damage measured using micronucleus frequency in human peripheral blood lymphocytes. Mutagenesis 2011, 26, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Fenech, M.; Morley, A.A. The effect of donor age on spontaneous and induced micronuclei. Mutat. Res. 1985, 148, 99–105. [Google Scholar] [CrossRef]
- Fenech, M.; Rinaldi, J. The relationship between micronuclei in human lymphocytes and plasma levels of vitamin C, vitamin E, vitamin B12 and folic acid. Carcinogenesis 1994, 15, 1405–1411. [Google Scholar] [CrossRef]
- Bonassi, S.; Neri, M.; Lando, C.; Ceppi, M.; Lin, Y.P.; Chang, W.P.; Holland, N.; Kirsch-Volders, M.; Zeiger, E.; Fenech, M.; et al. Effect of smoking habit on the frequency of micronuclei in human lymphocytes: Results from the Human MicroNucleus project. Mutat. Res. 2003, 543, 155–166. [Google Scholar] [CrossRef]
- Maffei, F.; Fimognari, C.; Castelli, E.; Stefanini, G.F.; Forti, G.C.; Hrelia, P. Increased cytogenetic damage detected by FISH analysis on micronuclei in peripheral lymphocytes from alcoholics. Mutagenesis 2000, 15, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Alvarenga, T.A.; Andersen, M.L.; Ribeiro, D.A.; Araujo, P.; Hirotsu, C.; Costa, J.L.; Battisti, M.C.; Tufik, S. Single exposure to cocaine or ecstasy induces DNA damage in brain and other organs of mice. Addict. Biol. 2010, 15, 96–99. [Google Scholar] [CrossRef]
- Schiffl, C.; Zieres, C.; Zankl, H. Exhaustive physical exercise increases frequency of micronuclei. Mutat. Res. 1997, 389, 243–246. [Google Scholar] [CrossRef]
- Mastaloudis, A.; Morrow, J.D.; Hopkins, D.W.; Devaraj, S.; Traber, M.G. Antioxidant supplementation prevents exercise-induced lipid peroxidation, but not inflammation, in ultramarathon runners. Free Radic. Biol. Med. 2004, 36, 1329–1341. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181–182, 219–222. [Google Scholar] [CrossRef]
- Ramana, K.V.; Reddy, A.B.M.; Majeti, N.; Singhal, S.S. Therapeutic Potential of Natural Antioxidants. Oxidative Med. Cell. Longev. 2018, 2018, 9471051. [Google Scholar] [CrossRef] [Green Version]
- Kouvaris, J.R.; Kouloulias, V.E.; Vlahos, L.J. Amifostine: The first selective-target and broad-spectrum radioprotector. Oncologist 2007, 12, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.A.; Kirkpatrick, D.R.; Smith, S.; Smith, T.K.; Pearson, T.; Kailasam, A.; Herrmann, K.Z.; Schubert, J.; Agrawal, D.K. Radioprotective agents to prevent cellular damage due to ionizing radiation. J. Transl. Med. 2017, 15, 232. [Google Scholar] [CrossRef]
- Kuefner, M.A.; Brand, M.; Ehrlich, J.; Braga, L.; Uder, M.; Semelka, R.C. Effect of antioxidants on X-ray-induced gamma-H2AX foci in human blood lymphocytes: Preliminary observations. Radiology 2012, 264, 59–67. [Google Scholar] [CrossRef]
- Coppernoll-Blach, P. Quertle: The Conceptual Relationships Alternative Search Engine for PubMed. J. Med. Libr. Assoc. 2011, 99, 176–177. [Google Scholar] [CrossRef]
- Cooke, M.S.; Patel, K.; Ahmad, J.; Holloway, K.; Evans, M.D.; Lunec, J. Monoclonal antibody to single-stranded DNA: A potential tool for DNA repair studies. Biochem. Biophys. Res. Commun. 2001, 284, 232–238. [Google Scholar] [CrossRef]
- Loo, D.T. In situ detection of apoptosis by the TUNEL assay: An overview of techniques. Methods Mol. Biol. 2011, 682, 3–13. [Google Scholar] [CrossRef]
- Holton, N.W.; Ebenstein, Y.; Gassman, N.R. Broad spectrum detection of DNA damage by Repair Assisted Damage Detection (RADD). DNA Repair (Amst.) 2018, 66–67, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Petrini, J.H. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003, 1, 207–218. [Google Scholar]
- Jurvansuu, J.; Raj, K.; Stasiak, A.; Beard, P. Viral Transport of DNA Damage That Mimics a Stalled Replication Fork. J. Virol. 2005, 79, 569–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaithamby, A.; Chen, D.J. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat. Res. 2011, 711, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [Green Version]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy—The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Lorat, Y.; Schanz, S.; Schuler, N.; Wennemuth, G.; Rube, C.; Rube, C.E. Beyond repair foci: DNA double-strand break repair in euchromatic and heterochromatic compartments analyzed by transmission electron microscopy. PLoS ONE 2012, 7, e38165. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rube, C.E. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef]
- Lorat, Y.; Schanz, S.; Rube, C.E. Ultrastructural Insights into the Biological Significance of Persisting DNA Damage Foci after Low Doses of Ionizing Radiation. Clin. Cancer Res. 2016, 22, 5300–5311. [Google Scholar] [CrossRef] [Green Version]
- Gustafsdottir, S.M.; Schallmeiner, E.; Fredriksson, S.; Gullberg, M.; Soderberg, O.; Jarvius, M.; Jarvius, J.; Howell, M.; Landegren, U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005, 345, 2–9. [Google Scholar] [CrossRef]
- Galbiati, A.; d’Adda di Fagagna, F. DNA Damage In Situ Ligation Followed by Proximity Ligation Assay (DI-PLA). Methods Mol. Biol. 2019, 1896, 11–20. [Google Scholar] [CrossRef]
- Novak, J.; Zamostna, B.; Vopalensky, V.; Buryskova, M.; Burysek, L.; Doleckova, D.; Pospisek, M. Interleukin-1α associates with the tumor suppressor p53 following DNA damage. Sci. Rep. 2020, 10, 6995. [Google Scholar] [CrossRef]
- Alsemarz, A.; Lasko, P.; Fagotto, F. Limited significance of the in situ proximity ligation assay. BioRxiv 2018, 411355. [Google Scholar] [CrossRef]
- Xu, R.; Yu, S.; Zhu, D.; Huang, X.; Xu, Y.; Lao, Y.; Tian, Y.; Zhang, J.; Tang, Z.; Zhang, Z.; et al. hCINAP regulates the DNA-damage response and mediates the resistance of acute myelocytic leukemia cells to therapy. Nat. Commun. 2019, 10, 3812. [Google Scholar] [CrossRef]
- Qin, P.W.; Parlak, M.; Kuscu, C.; Bandaria, J.; Mir, M.; Szlachta, K.; Singh, R.; Darzacq, X.; Yildiz, A.; Adli, M. Live cell imaging of low- and non-repetitive chromosome loci using CRISPR-Cas9. Nat. Commun. 2017, 8, 14725. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Georgescu, W.; Osseiran, A.; Rojec, M.; Liu, Y.; Bombrun, M.; Tang, J.; Costes, S.V. Characterizing the DNA Damage Response by Cell Tracking Algorithms and Cell Features Classification Using High-Content Time-Lapse Analysis. PLoS ONE 2015, 10, e0129438. [Google Scholar] [CrossRef]
- Jakob, B.; Dubiak-Szepietowska, M.; Janiel, E.; Schmidt, A.; Durante, M.; Taucher-Scholz, G. Differential Repair Protein Recruitment at Sites of Clustered and Isolated DNA Double-Strand Breaks Produced by High-Energy Heavy Ions. Sci. Rep. 2020, 10, 1443. [Google Scholar] [CrossRef]
- Li, C.; Tebo, A.G.; Gautier, A. Fluorogenic Labeling Strategies for Biological Imaging. Int. J. Mol. Sci. 2017, 18, 1473. [Google Scholar] [CrossRef] [Green Version]
- Bozhanova, N.G.; Baranov, M.S.; Klementieva, N.V.; Sarkisyan, K.S.; Gavrikov, A.S.; Yampolsky, I.V.; Zagaynova, E.V.; Lukyanov, S.A.; Lukyanov, K.A.; Mishin, A.S. Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal. Chem. Sci. 2017, 8, 7138–7142. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.K.; Ono, T.; Wang, S.; Jiang, W.; Franzini, R.M.; Jung, J.W.; Chan, K.M.; Kool, E.T. In Vitro Fluorogenic Real-Time Assay of the Repair of Oxidative DNA Damage. Chembiochem 2015, 16, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Beharry, A.A.; Nagel, Z.D.; Samson, L.D.; Kool, E.T. Fluorogenic Real-Time Reporters of DNA Repair by MGMT, a Clinical Predictor of Antitumor Drug Response. PLoS ONE 2016, 11, e0152684. [Google Scholar] [CrossRef] [Green Version]
- Beharry, A.A.; Lacoste, S.; O’Connor, T.R.; Kool, E.T. Fluorescence Monitoring of the Oxidative Repair of DNA Alkylation Damage by ALKBH3, a Prostate Cancer Marker. J. Am. Chem. Soc. 2016, 138, 3647–3650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.L.; Beharry, A.A.; Srivastava, A.; O’Connor, T.R.; Kool, E.T. Fluorescence Probes for ALKBH2 Allow the Measurement of DNA Alkylation Repair and Drug Resistance Responses. Angew. Chem. Int. Ed. Engl. 2018, 57, 12896–12900. [Google Scholar] [CrossRef]
- Wu, H.Y.; Cao, C.Y. The application of CRISPR-Cas9 genome editing tool in cancer immunotherapy. Brief. Funct. Genom. 2019, 18, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef] [Green Version]
- Mao, S.; Ying, Y.; Wu, X.; Krueger, C.J.; Chen, A.K. CRISPR/dual-FRET molecular beacon for sensitive live-cell imaging of non-repetitive genomic loci. Nucleic Acids Res. 2019, 47, e131. [Google Scholar] [CrossRef]
- Chen, B.; Zou, W.; Xu, H.; Liang, Y.; Huang, B. Efficient labeling and imaging of protein-coding genes in living cells using CRISPR-Tag. Nat. Commun. 2018, 9, 5065. [Google Scholar] [CrossRef]
- Wu, X.; Mao, S.; Ying, Y.; Krueger, C.J.; Chen, A.K. Progress and Challenges for Live-cell Imaging of Genomic Loci Using CRISPR-based Platforms. Genom. Proteom. Bioinform. 2019, 17, 119–128. [Google Scholar] [CrossRef]
- Sharma, A.; Toepfer, C.N.; Ward, T.; Wasson, L.; Agarwal, R.; Conner, D.A.; Hu, J.H.; Seidman, C.E. CRISPR/Cas9-Mediated Fluorescent Tagging of Endogenous Proteins in Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2018, 96, 21.11.1–21.11.20. [Google Scholar] [CrossRef]
- Bundell, S. CRISPR: Gene Editing and Beyond. Nature Video. Youtube. 2018. Available online: https://www.youtube.com/watch?v=4YKFw2KZA5o (accessed on 1 October 2020).
- Khan, A.O.; White, C.W.; Pike, J.A.; Yule, J.; Slater, A.; Hill, S.J.; Poulter, N.S.; Thomas, S.G.; Morgan, N.V. Optimised insert design for improved single-molecule imaging and quantification through CRISPR-Cas9 mediated knock-in. Sci. Rep. 2019, 9, 14219. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, V.; Mladenov, E.; Iliakis, G. Novel Biological Approaches for Testing the Contributions of Single DSBs and DSB Clusters to the Biological Effects of High LET Radiation. Front. Oncol. 2016, 6, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaithamby, A.; Chen, D.J. Cellular responses to DNA double-strand breaks after low-dose γ-irradiation. Nucleic Acids Res. 2009, 37, 3912–3923. [Google Scholar] [CrossRef] [Green Version]
- Tobias, F.; Lob, D.; Lengert, N.; Durante, M.; Drossel, B.; Taucher-Scholz, G.; Jakob, B. Spatiotemporal dynamics of early DNA damage response proteins on complex DNA lesions. PLoS ONE 2013, 8, e57953. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, N.I.; Brunton, H.; Watanabe, R.; Shrikhande, A.; Hirayama, R.; Matsufuji, N.; Fujimori, A.; Murakami, T.; Okayasu, R.; Jeggo, P.; et al. Visualisation of gammaH2AX foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS ONE 2013, 8, e70107. [Google Scholar] [CrossRef]
- Imrichova, T.; Hubackova, S.; Kucerova, A.; Kosla, J.; Bartek, J.; Hodny, Z.; Vasicova, P. Dynamic PML protein nucleolar associations with persistent DNA damage lesions in response to nucleolar stress and senescence-inducing stimuli. Aging 2019, 11, 7206–7235. [Google Scholar] [CrossRef]
- Varga, D.; Majoros, H.; Ujfaludi, Z.; Erdelyi, M.; Pankotai, T. Quantification of DNA damage induced repair focus formation via super-resolution dSTORM localization microscopy. Nanoscale 2019, 11, 14226–14236. [Google Scholar] [CrossRef] [Green Version]
- Blumhardt, P.; Stein, J.; Mucksch, J.; Stehr, F.; Bauer, J.; Jungmann, R.; Schwille, P. Photo-Induced Depletion of Binding Sites in DNA-PAINT Microscopy. Molecules 2018, 23, 3165. [Google Scholar] [CrossRef] [Green Version]
- Chow, T.T.; Shi, X.; Wei, J.-H.; Guan, J.; Stadler, G.; Huang, B.; Blackburn, E.H. Local enrichment of HP1alpha at telomeres alters their structure and regulation of telomere protection. Nat. Commun. 2018, 9, 3583. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, S.M.; Zhao, T.; Rozario, A.M.; Rootes, C.L.; McMillan, P.J.; Purcell, A.W.; Woon, A.; Marsh, G.A.; Lieu, K.G.; Wang, L.F.; et al. Viral regulation of host cell biology by hijacking of the nucleolar DNA-damage response. Nat. Commun. 2018, 9, 3057. [Google Scholar] [CrossRef] [Green Version]
- Hausmann, M.; Wagner, E.; Lee, J.H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuss, F.; Port, M.; Bestvater, F.; Scherthan, H. Super-resolution localization microscopy of radiation-induced histone H2AX-phosphorylation in relation to H3K9-trimethylation in HeLa cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef]
- Eryilmaz, M.; Schmitt, E.; Krufczik, M.; Theda, F.; Lee, J.H.; Cremer, C.; Bestvater, F.; Schaufler, W.; Hausmann, M.; Hildenbrand, G. Localization Microscopy Analyses of MRE11 Clusters in 3D-Conserved Cell Nuclei of Different Cell Lines. Cancers 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Horl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef]
- Betzig, E. Single Molecules, Cells, and Super-Resolution Optics (Nobel Lecture). Angew. Chem. Int. Ed. 2015, 54, 8034–8053. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Cricenti, A. Scanning Near-Field Optical Microscopy. In Encyclopedia of Condensed Matter Physics; Bassani, F., Liedl, G.L., Wyder, P., Eds.; Elsevier: Oxford, UK, 2005; pp. 163–171. [Google Scholar] [CrossRef]
- Qiu, Z.; Wilson, R.S.; Liu, Y.; R Dun, A.; Saleeb, R.S.; Liu, D.; Rickman, C.; Frame, M.; Duncan, R.R.; Lu, W. Translation Microscopy (TRAM) for super-resolution imaging. Sci. Rep. 2016, 6, 19993. [Google Scholar] [CrossRef] [Green Version]
- Varga, D.; Majoros, H.; Újfaludi, Z.; Erdélyi, M.; Pankotai, T. Quantification of DNA damage induced γH2AX focus formation via super-resolution dSTORM localization microscopy. BioRxiv 2019, 638361. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.; Krufczik, M.; Heermann, D.W.; Hausmann, M. Using Persistent Homology as a New Approach for Super-Resolution Localization Microscopy Data Analysis and Classification of γH2AX Foci/Clusters. Int. J. Mol. Sci. 2018, 19, 2263. [Google Scholar] [CrossRef] [Green Version]
- Liddle, P.; Jara-Wilde, J.; Lafon-Hughes, L.; Castro, I.; Härtel, S.; Folle, G. dSTORM microscopy evidences in HeLa cells clustered and scattered γH2AX nanofoci sensitive to ATM, DNA-PK, and ATR kinase inhibitors. Mol. Cell. Biochem. 2020, 473, 77–91. [Google Scholar] [CrossRef]
- Xu, X.; Nakano, T.; Tsuda, M.; Kanamoto, R.; Hirayama, R.; Uzawa, A.; Ide, H. Direct observation of damage clustering in irradiated DNA with atomic force microscopy. Nucleic Acids Res. 2019, 48, e18. [Google Scholar] [CrossRef]
- Tsuno, K. Resolution Limit of a Transmission Electron-Microscope with an Uncorrected Conventional Magnetic Objective Lens. Ultramicroscopy 1993, 50, 245–253. [Google Scholar] [CrossRef]
- Lorat, Y.; Fleckenstein, J.; Görlinger, P.; Rübe, C.; Rübe, C.E. Assessment of DNA damage by 53PB1 and pKu70 detection in peripheral blood lymphocytes by immunofluorescence and high-resolution transmission electron microscopy. Strahlenther. Onkol. 2020, 196, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Chen, B.K.; Liu, J.; Zhou, C.; Anchel, D.; Li, X.; Ge, J.; Bazett-Jones, D.P.; Sun, Y. Fluorescence and SEM correlative microscopy for nanomanipulation of subcellular structures. Light Sci. Appl. 2014, 3, e224. [Google Scholar] [CrossRef] [Green Version]
- SciencephotoLibrary. Cromosome SEM; Science Photo Library (SPL), Science Photo Library Limited: London, UK, 2020; Available online: https://www.sciencephoto.com/search?search=&q=chromosomes%20SEM&media_type=images (accessed on 1 October 2020).
- Jost, B.; Sergienko, A.; Abouraddy, A.; Saleh, B.; Teich, M. Spatial correlations of spontaneously down-converted photon pairs detected with a single-photon-sensitive CCD camera. Opt. Express 1998, 3, 81–88. [Google Scholar] [CrossRef]
- Lengert, N.; Mirsch, J.; Weimer, R.N.; Schumann, E.; Haub, P.; Drossel, B.; Lobrich, M. AutoFoci, an automated high-throughput foci detection approach for analyzing low-dose DNA double-strand break repair. Sci. Rep. 2018, 8, 17282. [Google Scholar] [CrossRef] [Green Version]
- Vincent, L. Morphological grayscale reconstruction in image analysis: Applications and efficient algorithms. IEEE Trans. Image Process. A Publ. IEEE Signal. Process. Soc. 1993, 2, 176–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivo-Marin, J.C. Extraction of spots in biological images using multiscale products. Pattern Recognit. 2002, 35, 1989–1996. [Google Scholar] [CrossRef]
- Neumaier, T.; Swenson, J.; Pham, C.; Polyzos, A.; Lo, A.T.; Yang, P.; Dyball, J.; Asaithamby, A.; Chen, D.J.; Bissell, M.J.; et al. Evidence for formation of DNA repair centers and dose-response nonlinearity in human cells. Proc. Natl. Acad. Sci. USA 2012, 109, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103 Pt 3, 857–862. [Google Scholar]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [Green Version]
- Demandolx, D.; Davoust, J. Subcellular Cytofluorometry in Confocal Microscopy. In Fluorescence Microscopy and Fluorescent Probes; Slavík, J., Ed.; Springer: Boston, MA, USA, 1996; pp. 279–283. [Google Scholar] [CrossRef]
- Martin, O.A.; Ivashkevich, A.; Choo, S.; Woodbine, L.; Jeggo, P.A.; Martin, R.F.; Lobachevsky, P. Statistical analysis of kinetics, distribution and co-localisation of DNA repair foci in irradiated cells: Cell cycle effect and implications for prediction of radiosensitivity. DNA Repair 2013, 12, 844–855. [Google Scholar] [CrossRef]
- Nikitaki, Z.; Nikolov, V.; Mavragani, I.V.; Plante, I.; Emfietzoglou, D.; Iliakis, G.; Georgakilas, A.G. Non-DSB clustered DNA lesions. Does theory colocalize with the experiment? Radiat. Phys. Chem. 2016, 128, 26–35. [Google Scholar] [CrossRef]
- Scherthan, H.; Lee, J.-H.; Maus, E.; Schumann, S.; Muhtadi, R.; Chojowski, R.; Port, M.; Lassmann, M.; Bestvater, F.; Hausmann, M. Nanostructure of Clustered DNA Damage in Leukocytes after In-Solution Irradiation with the Alpha Emitter Ra-223. Cancers 2019, 11, 1877. [Google Scholar] [CrossRef] [Green Version]
- Schubert, E.; Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X. DBSCAN Revisited, Revisited: Why and How You Should (Still) Use DBSCAN. ACM Trans. Database Syst. 2017, 42, 1–21. [Google Scholar] [CrossRef]
- Mascalchi, P.; Cordelières, F.P. Which Elements to Build Co-localization Workflows? From Metrology to Analysis. In Computer Optimized Microscopy: Methods and Protocols; Rebollo, E., Bosch, M., Eds.; Springer: New York, NY, USA, 2019; pp. 177–213. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Rudner, J.; Jendrossek, V. The Focinator—A new open-source tool for high-throughput foci evaluation of DNA damage. Radiat. Oncol. 2015, 10, 163. [Google Scholar] [CrossRef] [Green Version]
- Stauffer, W.; Sheng, H.; Lim, H.N. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 2018, 8, 15764. [Google Scholar] [CrossRef]
- Bray, M.A.; Carpenter, A.E. CellProfiler Tracer: Exploring and validating high-throughput, time-lapse microscopy image data. BMC Bioinform. 2015, 16, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Sauvat, A.; Leduc, M.; Müller, K.; Kepp, O.; Kroemer, G. ColocalizR: An open-source application for cell-based high-throughput colocalization analysis. Comput. Biol. Med. 2019, 107, 227–234. [Google Scholar] [CrossRef]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet. Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Ji, K.; Wang, Y.; Du, L.; Xu, C.; Liu, Y.; He, N.; Wang, J.; Liu, Q. Research Progress on the Biological Effects of Low-Dose Radiation in China. Dose-Response A Publ. Int. Hormesis Soc. 2019, 17, 1559325819833488. [Google Scholar] [CrossRef] [Green Version]
- Ermolaeva, M.A.; Segref, A.; Dakhovnik, A.; Ou, H.-L.; Schneider, J.I.; Utermöhlen, O.; Hoppe, T.; Schumacher, B. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 2013, 501, 416–420. [Google Scholar] [CrossRef] [Green Version]
- Martin, O.A.; Redon, C.E.; Nakamura, A.J.; Dickey, J.S.; Georgakilas, A.G.; Bonner, W.M. Systemic DNA Damage Related to Cancer. Cancer Res. 2011, 71, 3437. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.G.; Pefani, D.-E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Magnander, K.; Elmroth, K. Biological consequences of formation and repair of complex DNA damage. Cancer Lett. 2012, 327, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, R.; Rahmanian, S.; Nikjoo, H. Spectrum of Radiation-Induced Clustered Non-DSB Damage—A Monte Carlo Track Structure Modeling and Calculations. Radiat. Res. 2015, 183, 525–540. [Google Scholar] [CrossRef]
- Grudzenski, S.; Raths, A.; Conrad, S.; Rübe, C.E.; Löbrich, M. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 14205. [Google Scholar] [CrossRef] [Green Version]
- Löbrich, M.; Rief, N.; Kühne, M.; Heckmann, M.; Fleckenstein, J.; Rübe, C.; Uder, M. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc. Natl. Acad. Sci. USA 2005, 102, 8984–8989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, P.; Ishchenko, A.A.; Laval, J.; Paap, B.; Sutherland, B.M. Endogenous DNA damage clusters in human hematopoietic stem and progenitor cells. Free Radic. Biol. Med. 2008, 45, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Wukovich, R.L.; Aziz, K.; Kalogerinis, P.T.; Richardson, C.C.; Panayiotidis, M.I.; Bonner, W.M.; Sedelnikova, O.A.; Georgakilas, A.G. Accumulation of oxidatively induced clustered DNA lesions in human tumor tissues. Mutat. Res. 2009, 674, 131–136. [Google Scholar] [CrossRef]
- Ii, P.D.C.; Nakamura, J.; Rao, S.; Chu, H.; Ibrahim, J.G.; Swenberg, J.A.; Kaufman, D.G. Abasic sites preferentially form at regions undergoing DNA replication. FASEB J. 2010, 24, 3674–3680. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Collins, L.B.; Chen, T.H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative stress at low levels can induce clustered DNA lesions leading to NHEJ mediated mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Bibault, J.-E.; Tinhofer, I. The role of Next-Generation Sequencing in tumoral radiosensitivity prediction. Clin. Transl. Radiat. Oncol. 2017, 3, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Nikitaki, Z.; Hola, M.; Dona, M.; Pavlopoulou, A.; Michalopoulos, I.; Angelis, K.J.; Georgakilas, A.G.; Macovei, A.; Balestrazzi, A. Integrating plant and animal biology for the search of novel DNA damage biomarkers. Mutat Res. 2018, 775, 21–38. [Google Scholar] [CrossRef]
- Ventura, J.; Lobachevsky, P.N.; Palazzolo, J.S.; Forrester, H.; Haynes, N.M.; Ivashkevich, A.; Stevenson, A.W.; Hall, C.J.; Ntargaras, A.; Kotsaris, V.; et al. Localized Synchrotron Irradiation of Mouse Skin Induces Persistent Systemic Genotoxic and Immune Responses. Cancer Res. 2017, 77, 6389–6399. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Bi, K.; Yang, R.; Li, H.; Nikitaki, Z.; Chang, L. Role of DNA damage and repair in radiation cancer therapy: A current update and a look to the future. Int. J. Radiat. Biol. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | Question | Detection Method |
|---|---|---|
| tissue < 1 mm | Located in the same cell type? | Bright field microscopy |
| cellular > 10 μm | Located in the same cell? | BFM/Fluorescence microscopy |
| sub-cellular < 10 μm | Located in the same organelle of a cell? | Fluorescence microscopy/FRET |
| sub-light microscopic < 200 nm | Existence of a contact between proteins? | FRET/Electron microscopy STED nanoscopy |
| molecular < 1 nm | Point of contact between proteins? | Electron microscopy/AFM |
| Yr | Term | Meaning | Current Lateral Resolution | Current Axial Resolution | Wide Field or Point-Scanning | Single Molecule |
|---|---|---|---|---|---|---|
| 1931 | TEM | Transmission electron M. | 50 pm | 2 nm 1 | Point | Y |
| 1937 | SEM | Scanning electron M. | 0.4 nm | 2 nm 2 | Point | N |
| 1957 | CM | Confocal M. | 250 nm | 600 nm | Point | Ν |
| 1967 | LSC | Laser scanning confocal M. | 240 nm | 600 nm | Point | Ν |
| 1976 | FRAP | Fluorescence recovery after photobleaching | 250 nm | 1 µm | Both | N |
| 1981 | SPM | Scanning probe M. | 2 pm | 0.2 pm | Point | Y |
| 1982 | STM | Scanning tunneling M. | 0.1 nm | 10 pm | Point | Y |
| 1984 | SNOM | Scanning near field optical M. | <50 nm | 2 nm | Point | N |
| 1985 | AFM | Atomic Force M. | <1 nm | 0.2 pm | Point | Y |
| 1990 | 2PEF | 2 Photon excitation fluorescence M. | 300 nm | 500 nm | Point | N |
| 1990 | PSTM | Photon scanning tunneling M. | 10 pm | 0.3 pm | Point | Y |
| 1993 | FCS | Fluorescence correlation spectroscopy | 1 µm | 1 µm | Point | N |
| 2006 | STORM | Stochastic optical reconstruction M. | <30 nm | 10 nm | Wide | Y |
| 2006 | PALM | Photoactivated localization M. | 10 nm | 10 nm | Wide | Y |
| 2008 | 3D SIM | 3D Structured illumination M. | 100 nm | 250 nm | Wide | N |
| 2009 | SPIM or LSM (LSFM) | Selective plane illumination M. Light sheet (fl.) M | 300 nm | 800 nm | Wide | N |
| 2010 | Bessel LSM | Bessel light sheet M. | 300 nm | 800 nm | Wide | N |
| 2014 | Lattice LSM | Lattice light sheet M. | 75 nm | 100 nm | Wide | N |
| 2015 | STED nanoscopy | Stimulated Emission Depletion | 80 nm | 100 nm | Point | Y |
| 2016 | TRAM | Translation M. | 7-fold res improv. | 7-fold | Wide | N |
| 2016 | SRRF | Super-resolution Radial Fluctuations | Depends on microscopy | Both | Y | |
| # | Name | Objective | Concept | Ref. |
|---|---|---|---|---|
| 1 | Local Maximum (algorithm) | Find the pixel(s) with maximum intensity in a delimited area. | Intensity comparison of neighboring pixels to select the pixel(s) with the higher intensity value | |
| 2 | Intensity threshold (criterion) | Distinguish the signal from the background to define the boundary of a (convex) object. | Intensity comparison of neighboring pixels; which edge pixels should be discarded as noise and which should be retained. | |
| 3 | Area growing (algorithm) | a Define the boundary of a (convex) object. b Distinguish signal from the background. | a The area is defined from a pixel of local maximum intensity (seed) and continuing including neighboring pixels until reaching a specified intensity threshold value. b Same process beginning from the local minimum intensity pixel(s). | [120] |
| 4 | (white) Top hat (transformation) | Extract small elements or details of an image. | Selects objects that are smaller than a “structuring element” 1 and brighter than their surroundings | |
| 5 | H-dome (transformation) | Extract small elements or details of an image. | Excludes background by keeping local maxima above an intensity threshold defined from the local background, rescaling the intensity values by subtracting local background levels of each “dome.” | [121] |
| 6 | A Trous Wavelet | Enhance contrast in the image by reducing noise from non-specific signals using pattern recognition algorithm. | a Creates an intensity-independent image. b Applies a constant threshold on the wavelet filtered image. c Watershed algorithms separate adjacent spots, based on the aminimum size threshold. | [122,123] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikitaki, Z.; Pariset, E.; Sudar, D.; Costes, S.V.; Georgakilas, A.G. In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead. Cancers 2020, 12, 3288. https://doi.org/10.3390/cancers12113288
Nikitaki Z, Pariset E, Sudar D, Costes SV, Georgakilas AG. In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead. Cancers. 2020; 12(11):3288. https://doi.org/10.3390/cancers12113288
Chicago/Turabian StyleNikitaki, Zacharenia, Eloise Pariset, Damir Sudar, Sylvain V. Costes, and Alexandros G. Georgakilas. 2020. "In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead" Cancers 12, no. 11: 3288. https://doi.org/10.3390/cancers12113288
APA StyleNikitaki, Z., Pariset, E., Sudar, D., Costes, S. V., & Georgakilas, A. G. (2020). In Situ Detection of Complex DNA Damage Using Microscopy: A Rough Road Ahead. Cancers, 12(11), 3288. https://doi.org/10.3390/cancers12113288