DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer


Abstract
:Simple Summary
Abstract
1. Prologue: Mutations in DNA Pol Genes and Cancer
2. Loss of Replication Fidelity Control Elevates Mutation Rates: Classic Rules
3. The Cornerstone Model of the Replication Fork
4. Progress on the Structure-Function of B-Family DNA Polymerases and Organization of the Replication Fork
5. DNA Polymerase Genes Mutations in Cancer
6. Conclusions: A Projection into the Future
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rayner, E.; van Gool, I.C.; Palles, C.; Kearsey, S.E.; Bosse, T.; Tomlinson, I.; Church, D.N. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat. Rev. Cancer 2016, 16, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Barbari, S.R.; Shcherbakova, P.V. Replicative DNA polymerase defects in human cancers: Consequences, mechanisms, and implications for therapy. DNA Repair 2017, 56, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA replication fidelity and cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahirov, T.H.; Makarova, K.S.; Rogozin, I.B.; Pavlov, Y.I.; Koonin, E.V. Evolution of DNA polymerases: An inactivated polymerase-exonuclease module in Pol epsilon and a chimeric origin of eukaryotic polymerases from two classes of archaeal ancestors. Biol. Direct 2009, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, D.; Krupovic, M.; Guglielmini, J.; Forterre, P.; Venclovas, Č. Diversity and evolution of B-family DNA polymerases. Nucleic Acids Res. 2020, 48, 10142–10156. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P. An attempt to unify the structure of polymerases. Protein Eng. 1990, 3, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Blanco, L.; Bernad, A.; Blasco, M.A.; Salas, M. A general structure for DNA-dependent DNA polymerases. Gene 1991, 100, 27–38. [Google Scholar] [CrossRef]
- Brautigam, C.A.; Steitz, T.A. Structural and functional insights provided by crystal structures of DNA polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1998, 8, 54–63. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Eichinger, A.; Engh, R.A.; Laue, F.; Ankenbauer, W.; Huber, R.; Angerer, B. Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc. Natl. Acad. Sci. USA 1999, 96, 3600–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kähler, M.; Antranikian, G. Cloning and characterization of a family B DNA polymerase from the hyperthermophilic crenarchaeon Pyrobaculum islandicum. J. Bacteriol. 2000, 182, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shevelev, I.V.; Hübscher, U. The 3′ 50′ exonucleases. Nat. Rev. Mol. Cell. Biol. 2002, 3, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Schaaper, R.M. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J. Biol. Chem. 1993, 268, 23762–23765. [Google Scholar] [PubMed]
- Kunkel, T.A. DNA replication fidelity. J. Biol. Chem. 2004, 279, 16895–16898. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, M.; Limsirichaikul, S.; Niimi, A.; Iwai, S.; Yoshida, S.; Suzuki, M. Distinct function of conserved amino acids in the fingers of Saccharomyces cerevisiae DNA polymerase α. J. Biol. Chem. 2003, 278, 19071–19078. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Murphy, K.M.; Kanevets, U.; Reha-Krantz, L.J. Sensitivity to phosphonoacetic acid: A new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics 2005, 170, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, R.N.; Hsu, J.J.; Lawrence, N.A.; Preston, B.D.; Loeb, L.A. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase δ. J. Biol. Chem. 2006, 281, 4486–4494. [Google Scholar] [CrossRef] [Green Version]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Regulation of B family DNA polymerase fidelity by a conserved active site residue: Characterization of M644W, M644L and M644F mutants of yeast DNA polymerase epsilon. Nucleic Αcids Res. 2007, 35, 3076–3086. [Google Scholar] [CrossRef]
- Sakamoto, A.N.; Stone, J.E.; Kissling, G.E.; McCulloch, S.D.; Pavlov, Y.I.; Kunkel, T.A. Mutator alleles of yeast DNA polymerase zeta. DNA Repair 2007, 6, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.; Bell, J.B.; Kunkel, T.A.; Sugino, A. Eukaryotic DNA polymerase amino acid sequence required for 3’→5’ exonuclease activity. Proc. Natl. Acad. Sci. USA 1991, 88, 9473–9477. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Obert, R.; Burgers, P.M.; Kunkel, T.A.; Resnick, M.A.; Gordenin, D.A. The 3′→5′ exonuclease of DNA polymerase δ can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc. Natl. Acad. Sci. USA 2001, 98, 5122–5127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Schmeits, J.L.; Amin, N.S.; Lau, P.J.; Myung, K.; Kolodner, R.D. Checkpoint-dependent activation of mutagenic repair in Saccharomyces cerevisiae pol3-01 mutants. Mol. Cell 2000, 6, 593–603. [Google Scholar] [CrossRef]
- Morrison, A.; Sugino, A. The 3′→5′ exonucleases of both DNA polymerases δ and ε participate in correcting errors of DNA replication in Saccharomyces cerevisiae. Mol. Gen. Genet. 1994, 242, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. DNA polymerase delta proofreads errors made by DNA polymerase epsilon. Proc. Natl. Acad. Sci. USA 2020, 117, 6035–6041. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, Y.I.; Frahm, C.; McElhinny, S.A.; Niimi, A.; Suzuki, M.; Kunkel, T.A. Evidence that errors made by DNA polymerase α are corrected by DNA polymerase δ. Curr. Biol. 2006, 16, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nick McElhinny, S.A.; Kissling, G.E.; Kunkel, T.A. Differential correction of lagging-strand replication errors made by DNA polymerases α and δ. Proc. Natl. Acad. Sci. USA 2010, 107, 21070–21075. [Google Scholar] [CrossRef] [Green Version]
- Herr, A.J.; Kennedy, S.R.; Knowels, G.M.; Schultz, E.M.; Preston, B.D. DNA replication error-induced extinction of diploid yeast. Genetics 2014, 196, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.; Araki, H.; Clark, A.B.; Hamatake, R.K.; Sugino, A. A third essential DNA polymerase in S. cerevisiae. Cell 1990, 62, 1143–1151. [Google Scholar] [CrossRef]
- Syväoja, J.; Suomensaari, S.; Nishida, C.; Goldsmith, J.S.; Chui, G.S.; Jain, S.; Linn, S. DNA polymerases alpha, delta, and epsilon: Three distinct enzymes from HeLa cells. Proc. Natl. Acad. Sci. USA 1990, 87, 6664–6668. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Kao, H.I.; Bambara, R.A. Flap endonuclease 1: A central component of DNA metabolism. Annu. Rev. Biochem. 2004, 73, 589–615. [Google Scholar] [CrossRef] [PubMed]
- Reijns, M.A.M.; Kemp, H.; Ding, J.; de Procé, S.M.; Jackson, A.P.; Taylor, M.S. Lagging-strand replication shapes the mutational landscape of the genome. Nature 2015, 518, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.X.; Lujan, S.A.; Burkholder, A.B.; Garbacz, M.A.; Kunkel, T.A. Roles for DNA polymerase δ in initiating and terminating leading strand DNA replication. Nat. Commun. 2019, 10, 3992. [Google Scholar] [CrossRef] [Green Version]
- Nick McElhinny, S.A.; Gordenin, D.A.; Stith, C.M.; Burgers, P.M.; Kunkel, T.A. Division of labor at the eukaryotic replication fork. Mol. Cell 2008, 30, 137–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliam, T.A.; Yeeles, J.T.P. An updated perspective on the polymerase division of labor during eukaryotic DNA replication. Crit Rev. Biochem. Mol. Biol. 2020, 55, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Shcherbakova, P.V.; Pavlov, Y.I. 3′→5′ exonucleases of DNA polymerases ε and δ correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics 1996, 142, 717–726. [Google Scholar] [PubMed]
- Karthikeyan, R.; Vonarx, E.J.; Straffon, A.F.; Simon, M.; Faye, G.; Kunz, B.A. Evidence from mutational specificity studies that yeast DNA polymerases δ and ε replicate different DNA strands at an intracellular replication fork. J. Mol. Biol. 2000, 299, 405–419. [Google Scholar] [CrossRef]
- Kunkel, T.A. Biological asymmetries and the fidelity of eukaryotic DNA replication. Bioessays 1992, 14, 303–308. [Google Scholar] [CrossRef]
- Waga, S.; Stillman, B. Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature 1994, 369, 207–212. [Google Scholar] [CrossRef]
- Burgers, P.M. Eukaryotic DNA polymerases in DNA replication and DNA repair. Chromosoma 1998, 107, 218–227. [Google Scholar] [CrossRef]
- Johnson, A.; O’Donnell, M. Cellular DNA replicases: Components and dynamics at the replication fork. Annu. Rev. Biochem. 2005, 74, 283–315. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Burgers, P.M. Dividing the workload at a eukaryotic replication fork. Trends Cell. Biol. 2008, 18, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA replication fork. Annu Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Pavlov, Y.I.; Shcherbakova, P.V. DNA polymerases at the eukaryotic fork-20 years later. Mutat. Res. 2010, 685, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aria, V.; Yeeles, J.T.P. Mechanism ofbidirectional leading-strand synthesis establishment at eukaryotic DNA replication origins. Mol. Cell 2018, 73, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, A.R.; Lujan, S.A.; Burkholder, A.B.; Orebaugh, C.D.; Williams, J.S.; Clausen, M.F.; Maiv, E.; Mieczkowski, P.A.; Fargo, D.C.; Smith, D.J.; et al. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol. 2015, 22, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Guilliam, T.A.; Yeeles, J.T.P. Reconstitution of translesion synthesis reveals a mechanism of eukaryotic DNA replication restart. Nat. Struct. Mol. Biol. 2020, 27, 450–460. [Google Scholar] [CrossRef]
- Flood, C.L.; Rodriguez, G.P.; Bao, G.; Shockley, A.H.; Kow, Y.W.; Crouse, G.F. Replicative DNA polymerase δ but not ε proofreads errors in Cis and in Trans. PLoS Genet. 2015, 11, e1005049. [Google Scholar] [CrossRef] [Green Version]
- Kesti, T.; Flick, K.; Keranen, S.; Syvaoja, J.E.; Wittenberg, C. DNA polymerase ε catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol. Cell 1999, 3, 679–685. [Google Scholar] [CrossRef]
- Dua, R.; Levy, D.L.; Campbell, J.L. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol ε and its unexpected ability to support growth in the absence of the DNA polymerase domain. J. Biol. Chem. 1999, 274, 22283–22288. [Google Scholar] [CrossRef] [Green Version]
- Ohya, T.; Kawasaki, Y.; Hiraga, S.; Kanbara, S.; Nakajo, K.; Nakashima, N.; Suzuki, A.; Sugino, A. The DNA polymerase domain of pol(ε) is required for rapid, efficient, and highly accurate chromosomal DNA replication, telomere length maintenance, and normal cell senescence in Saccharomyces cerevisiae. J. Biol. Chem. 2002, 277, 28099–28108. [Google Scholar] [CrossRef] [Green Version]
- Garbacz, M.A.; Lujan, S.A.; Burkholder, A.B.; Cox, P.B.; Wu, Q.; Zhou, Z.X.; Haber, J.E.; Kunkel, T.A. Evidence that DNA polymerase delta contributes to initiating leading strand DNA replication in Saccharomyces cerevisiae. Nat. Commun. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnianni, R.A.; Zhou, Z.X.; Lujan, S.A.; Al-Zain, A.; Garcia, V.; Glancy, E.; Burkholder, A.B.; Kunkel, T.A.; Symington, L.S. DNA polymerase delta synthesizes both strands during break-induced replication. Mol. Cell 2019, 76, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Stepchenkova, E.I.; Zhuk, A.S.; Cui, J.; Tarakhovskaya, E.R.; Barbari, S.R.; Shcherbakova, P.V.; Polev, D.E.; Fedorov, R.; Poliakov, E.; Rogozin, I.B.; et al. Compensation for the absence of the catalytically active half of DNA polymerase ε in yeast by positively selected mutations in CDC28 gene. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sviderskiy, V.O.; Blumenberg, L.; Gorodetsky, E.; Karakousi, T.R.; Hirsh, N.; Alvarez, S.W.; Terzi, E.M.; Kaparos, E.; Whiten, G.C.; Ssebyala, S.; et al. Hyperactive CDK2 activity in basal-like breast cancer imposes a genome integrity liability that can be exploited by targeting DNA polymerase ε. Mol. Cell 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L. The Pol alpha-Primase Complex. Subcell. Biochem. 2012, 62, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Agarkar, V.B.; Babayeva, N.D.; Pavlov, Y.I.; Tahirov, T.H. Crystal structure of the C-terminal domain of human DNA primase large subunit: Implications for the mechanism of the primase-polymerase α switch. Cell Cycle 2011, 10, 926–931. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Gu, J.; Suwa, Y.; Pavlov, Y.I.; Tahirov, T.H. Mechanism of concerted rna-dna primer synthesis by the human primosome. J. Biol. Chem. 2016, 291, 10006–10020. [Google Scholar] [CrossRef] [Green Version]
- Swan, M.K.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol. 2009, 16, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Rice, W.J.; Malik, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Cryo-EM structure and dynamics of eukaryotic DNA polymerase δ holoenzyme. Nat. Struct. Mol. Biol. 2019, 26, 955–962. [Google Scholar] [CrossRef]
- Lancey, C.; Tehseen, M.; Raducanu, V.S.; Rashid, F.; Merino, N.; Ragan, T.J.; Savva, C.G.; Zaher, M.S.; Shirbini, A.; Blanco, F.J.; et al. Structure of the processive human Pol δ holoenzyme. Nat. Commun. 2020, 11, 1109. [Google Scholar] [CrossRef] [Green Version]
- Hogg, M.; Osterman, P.; Bylund, G.O.; Ganai, R.A.; Lundstrom, E.B.; Sauer-Eriksson, A.E.; Johansson, E. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat. Struct. Mol. Biol. 2014, 21, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Rajashankar, K.R.; Buku, A.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Crystal structure of yeast DNA polymerase epsilon catalytic domain. PLoS ONE 2014, 9, e94835. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, R.; Yuan, Z.; Bai, L.; de Luna Almeida Santos, R.; Sun, J.; Zhang, D.; Yurieva, O.; Li, H.; O’Donnell, M.E. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc. Natl. Acad. Sci. USA 2017, 114, E697–E706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Georgescu, R.; Schauer, G.D.; O’Donnell, M.E.; Li, H. Structure of the polymerase ε holoenzyme and atomic model of the leading strand replisome. Nat. Commun. 2020, 11, 3156. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Kopylov, M.; Gomez-Llorente, Y.; Jain, R.; Johnson, R.E.; Prakash, L.; Prakash, S.; Ubarretxena-Belandia, I.; Aggarwal, A.K. Structure and mechanism of B-family DNA polymerase ζ specialized for translesion DNA synthesis. Nat. Struct. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Goswami, P.; Abid Ali, F.; Douglas, M.E.; Locke, J.; Purkiss, A.; Janska, A.; Eickhoff, P.; Early, A.; Nans, A.; Cheung, A.M.C.; et al. Structure of DNA-CMG-Pol epsilon elucidates the roles of the non-catalytic polymerase modules in the eukaryotic replisome. Nat. Commun. 2018, 9, 5061. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Zhang, S.; Wang, X.; Chao, H.H.; Zhao, H.; Darzynkiewicz, Z.; Zhang, Z.; Lee, E.Y.C. Two forms of human DNA polymerase δ: Who does what and why? DNA Repair 2019, 81, 102656. [Google Scholar] [CrossRef]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2012, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Siebler, H.M.; Pavlov, Y.I.; Tahirov, T.H. Iron-Sulfur clusters in DNA polymerases and primases of eukaryotes. Methods Enzymol. 2018, 599, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. The indispensable role of mammalian iron sulfur proteins in function and regulation of multiple diverse metabolic pathways. Biometals 2019, 32, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, E.J.; Boal, A.K.; Barton, J.K. Biological contexts for DNA charge transport chemistry. Curr. Opin. Chem. Biol. 2008, 12, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, S.; Hirst, J.; Maman, J.D.; Krude, T.; Pellegrini, L. An iron-sulfur domain of the eukaryotic primase is essential for RNA primer synthesis. Nat. Struct. Mol. Biol. 2007, 14, 875–877. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Zhang, Y.; Suwa, Y.; Gu, J.; Babayeva, N.D.; Pavlov, Y.I.; Tahirov, T.H. Insight into the Human DNA Primase Interaction with Template-Primer. J. Biol. Chem. 2016, 291, 4793–4802. [Google Scholar] [CrossRef] [Green Version]
- Holt, M.E.; Salay, L.E.; Chazin, W.J. A polymerase with potential: The Fe-S cluster in human DNA primase. Methods Enzymol. 2017, 595, 361–390. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Blanco, L.; Pavlov, Y.I.; Tahirov, T.H. Comment on The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Lada, A.G.; Siebler, H.M.; Zhang, Y.; Pavlov, Y.I.; Tahirov, T.H. DNA Polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J. Biol. Chem. 2012, 287, 17281–17287. [Google Scholar] [CrossRef] [Green Version]
- Bartels, P.L.; Stodola, J.L.; Burgers, P.M.J.; Barton, J.K. A Redox role for the [4Fe4S] cluster of yeast DNA polymerase δ. J. Am. Chem. Soc. 2017, 139, 18339–18348. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Vanamee, E.S.; Dzikovski, B.G.; Buku, A.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. An iron-sulfur cluster in the polymerase domain of yeast DNA polymerase epsilon. J. Mol. Biol. 2014, 426, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Ter Beek, J.; Parkash, V.; Bylund, G.O.; Osterman, P.; Sauer-Eriksson, A.E.; Johansson, E. Structural evidence for an essential Fe-S cluster in the catalytic core domain of DNA polymerase ε. Nucleic Acids Res. 2019, 47, 5712–5722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlov, Y.I.; Shcherbakova, P.V.; Rogozin, I.B. Roles of DNA polymerases in replication, repair, and recombination in eukaryotes. Int. Rev. Cytol. 2006, 255, 41–132. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.A.; Vassel, F.M.; Chatterjee, N.; D’Souza, S.; Li, Y.; Hao, B.; Hemann, M.T.; Walker, G.C.; Korzhnev, D.M. Rev7 dimerization is important for assembly and function of the Rev1/Polζ translesion synthesis complex. Proc. Natl. Acad. Sci. USA 2018, 115, E8191–E8200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.R.; Lawrence, C.W.; Hinkle, D.C. Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science 1996, 272, 1646–1649. [Google Scholar] [CrossRef]
- Haracska, L.; Unk, I.; Johnson, R.E.; Johansson, E.; Burgers, P.M.; Prakash, S.; Prakash, L. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001, 15, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Stith, C.M.; Majka, J.; Burgers, P.M. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase zeta. J. Biol. Chem. 2005, 280, 23446–23450. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.E.; Prakash, L.; Prakash, S. Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc. Natl. Acad. Sci. USA 2012, 109, 12455–12460. [Google Scholar] [CrossRef] [Green Version]
- Makarova, A.V.; Stodola, J.L.; Burgers, P.M. A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res. 2012, 40, 11618–11626. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Gregory, M.T.; Yang, W. Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc. Natl. Acad. Sci. USA. 2014, 111, 2954–2959. [Google Scholar] [CrossRef] [Green Version]
- Giot, L.; Chanet, R.; Simon, M.; Facca, C.; Faye, G. Involvement of the yeast DNA polymerase δ in DNA repair in vivo. Genetics 1997, 146, 1239–1251. [Google Scholar]
- Stepchenkova, E.I.; Tarakhovskaya, E.R.; Siebler, H.M.; Pavlov, Y.I. Defect of Fe-S cluster binding by DNA polymerase delta in yeast suppresses UV-induced mutagenesis, but enhances DNA polymerase zeta- dependent spontaneous mutagenesis. DNA Repair 2017, 49, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Pagès, V.; Johnson, R.E.; Prakash, L.; Prakash, S. Mutational specificity and genetic control of replicative bypass of an abasic site in yeast. Proc. Natl. Acad. Sci. USA 2008, 105, 1170–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, A.C.; Zhou, J.C.; Perera, R.L.; van Deursen, F.; Evrin, C.; Ivanova, M.E.; Kilkenny, M.L.; Renault, L.; Kjaer, S.; Matak-Vinković, D.; et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature 2014, 510, 293–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilkenny, M.L.; Simon, A.C.; Mainwaring, J.; Wirthensohn, D.; Holzer, S.; Pellegrini, L. The human CTF4-orthologue AND-1 interacts with DNA polymerase alpha/primase via its unique C-terminal HMG box. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Georgescu, R.; Santos, R.L.A.; Zhang, D.; Bai, L.; Yao, N.Y.; Zhao, G.; O’Donnell, M.E.; Li, H. Ctf4 organizes sister replisomes and Pol α into a replication factory. Elife 2019, 8. [Google Scholar] [CrossRef]
- Rzechorzek, N.J.; Hardwick, S.W.; Jatikusumo, V.A.; Chirgadze, D.Y.; Pellegrini, L. CryoEM structures of human CMG-ATPγS-DNA and CMG-AND-1 complexes. Nucleic Acids Res. 2020, 48, 6980–6995. [Google Scholar] [CrossRef]
- Huang, M.E.; Le Douarin, B.; Henry, C.; Galibert, F. The Saccharomyces cerevisiae protein YJR043C (Pol32) interacts with the catalytic subunit of DNA polymerase alpha and is required for cell cycle progression in G2/M. Mol. Gen. Genet. 1999, 260, 541–550. [Google Scholar] [CrossRef]
- Johansson, E.; Garg, P.; Burgers, P.M. The Pol32 subunit of DNA polymerase delta contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J. Biol. Chem. 2004, 279, 1907–1915. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Spenkelink, L.M.; Schauer, G.D.; Yurieva, O.; Mueller, S.H.; Natarajan, V.; Kaur, G.; Maher, C.; Kay, C.; O’Donnell, M.E.; et al. Tunability of DNA polymerase stability during eukaryotic DNA replication. Mol. Cell 2020, 77, 17–25. [Google Scholar] [CrossRef]
- Northam, M.R.; Moore, E.A.; Mertz, T.M.; Binz, S.K.; Stith, C.M.; Stepchenkova, E.I.; Wendt, K.L.; Burgers, P.M.; Shcherbakova, P.V. DNA polymerases ζ and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Res. 2014, 42, 290–306. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, Y.I.; Shcherbakova, P.V.; Kunkel, T.A. In vivo consequences of putative active site missense mutations in yeast replicative DNA polymerases α, ε, δ and ζ. Genetics 2001, 159, 47–64. [Google Scholar] [PubMed]
- Garbacz, M.; Araki, H.; Flis, K.; Bebenek, A.; Zawada, A.E.; Jonczyk, P.; Makiela-Dzbenska, K.; Fijalkowska, I.J. Fidelity consequences of the impaired interaction between DNA polymerase epsilon and the GINS complex. DNA Repair 2015, 29, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northam, M.R.; Garg, P.; Baitin, D.M.; Burgers, P.M.; Shcherbakova, P.V. A novel function of DNA polymerase zeta regulated by PCNA. EMBO J. 2006, 25, 4316–4325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.R.; Nguyen, H.D.; Wang, X.; Bielinsky, A.K. Mcm10 deficiency causes defective-replisome-induced mutagenesis and a dependency on error-free postreplicative repair. Cell Cycle 2014, 13, 1737–1748. [Google Scholar] [CrossRef] [Green Version]
- Kochenova, O.V.; Daee, D.L.; Mertz, T.M.; Shcherbakova, P.V. DNA polymerase ζ-dependent lesion bypass in Saccharomyces cerevisiae is accompanied by error-prone copying of long stretches of adjacent DNA. PLoS Genet. 2015, 11, e1005110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Whitehouse, I. Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 2012, 483, 434–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriramachandran, A.M.; Petrosino, G.; Méndez-Lago, M.; Schäfer, A.J.; Batista-Nascimento, L.S.; Zilio, N.; Ulrich, H.D. Genome-wide Nucleotide-Resolution Mapping of DNA Replication Patterns, Single-Strand Breaks, and Lesions by GLOE-Seq. Mol. Cell 2020, 78, 975–985. [Google Scholar] [CrossRef]
- Waisertreiger, I.S.; Liston, V.G.; Menezes, M.R.; Kim, H.M.; Lobachev, K.S.; Stepchenkova, E.I.; Tahirov, T.H.; Rogozin, I.B.; Pavlov, Y.I. Modulation of mutagenesis in eukaryotes by DNA replication fork dynamics and quality of nucleotide pools. Environ. Mol. Mutagen. 2012, 53, 699–724. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, Y.I.; Mian, I.M.; Kunkel, T.A. Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr. Biol. 2003, 13, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Hoopes, J.I.; Cortez, L.M.; Mertz, T.M.; Malc, E.P.; Mieczkowski, P.A.; Roberts, S.A. APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep. 2016, 14, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, A.S.; Hao, W.; Townes, J.P.; Lee, H.; Tang, H.; Foster, P.L. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. USA 2016, 113, 2176–2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronan, G.E.; Kouzminova, E.A.; Kuzminov, A. Near-continuously synthesized leading strands in Escherichia coli are broken by ribonucleotide excision. Proc. Natl. Acad. Sci. USA 2019, 116, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgers, P.M. Solution to the 50-year-old Okazaki-fragment problem. Proc. Natl. Acad. Sci. USA 2019, 116, 3358–3360. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Peltomäki, P. Lynch syndrome genes. Fam. Cancer 2005, 4, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [Green Version]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Mitchell, R.J.; Farrington, S.M.; Dunlop, M.G.; Campbell, H. Mismatch repair genes hMLH1 and hMSH2 and colorectal cancer: A HuGE review. Am. J. Epidemiol. 2002, 156, 885–902. [Google Scholar] [CrossRef] [PubMed]
- Galati, M.A.; Hodel, K.P.; Gams, M.S.; Sudhaman, S.; Bridge, T.; Zahurancik, W.J.; Ungerleider, N.A.; Park, V.S.; Ercan, A.B.; Joksimovic, L.; et al. Cancers from novel Pole mutant mouse models provide insights into polymerase-mediated hypermutagenesis and immune checkpoint blockade. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Church, D.N.; Briggs, S.E.; Palles, C.; Domingo, E.; Kearsey, S.J.; Grimes, J.M.; Gorman, M.; Martin, L.; Howarth, K.M.; Hodgson, S.V.; et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum. Mol. Genet. 2013, 22, 2820–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, V.S.; Pursell, Z.F. POLE proofreading defects: Contributions to mutagenesis and cancer. DNA Repair 2019, 76, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Kane, D.P.; Bulock, C.R.; Moore, E.A.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. A recurrent cancer-associated substitution in DNA polymerase ε produces a hyperactive enzyme. Nat. Commun. 2019, 10, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, D.P.; Shcherbakova, P.V. A common cancer-associated DNA polymerase ε mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014, 74, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrea, A.A.; Lujan, S.A.; Nick McElhinny, S.A.; Mieczkowski, P.A.; Resnick, M.A.; Gordenin, D.A.; Kunkel, T.A. Genome-wide model for the normal eukaryotic DNA replication fork. Proc. Natl. Acad. Sci. USA 2010, 107, 17674–17679. [Google Scholar] [CrossRef] [Green Version]
- Bulock, C.R.; Xing, X.; Shcherbakova, P.V. Mismatch repair and DNA polymerase δ proofreading prevent catastrophic accumulation of leading strand errors in cells expressing a cancer-associated DNA polymerase ϵ variant. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Parkash, V.; Kulkarni, Y.; Ter Beek, J.; Shcherbakova, P.V.; Kamerlin, S.C.L.; Johansson, E. Structural consequence of the most frequently recurring cancer-associated substitution in DNA polymerase ε. Nat. Commun. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Reha-Krantz, L.J. Learning about DNA polymerase function by studying antimutator DNA polymerases. Trends Biochem. Sci. 1995, 20, 136–140. [Google Scholar] [CrossRef]
- Beechem, J.M.; Otto, M.R.; Bloom, L.B.; Eritja, R.; Reha-Krantz, L.J.; Goodman, M.F. Exonuclease-polymerase active site partitioning of primer-template DNA strands and equilibrium Mg2+ binding properties of bacteriophage T4 DNA polymerase. Biochemistry 1998, 37, 10144–10155. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Darmawan, H.; Schultz, A.; Fidalgo da Silva, E.; Reha-Krantz, L.J. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome 2006, 49, 403–410. [Google Scholar] [CrossRef]
- Shcherbakova, P.V.; Pavlov, Y.I.; Chilkova, O.; Rogozin, I.B.; Johansson, E.; Kunkel, T.A. Unique error signature of the four-subunit yeast DNA polymerase epsilon. J. Biol. Chem. 2003, 278, 43770–43780. [Google Scholar] [CrossRef] [Green Version]
- Fortune, J.M.; Pavlov, Y.I.; Welch, C.M.; Johansson, E.; Burgers, P.M.; Kunkel, T.A. Saccharomyces cerevisiae DNA polymerase d: High fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J. Biol. Chem. 2005, 280, 29980–29987. [Google Scholar] [CrossRef] [Green Version]
- Li, H.D.; Cuevas, I.; Zhang, M.; Lu, C.; Alam, M.M.; Fu, Y.X.; You, M.J.; Akbay, E.A.; Zhang, H.; Castrillon, D.H. Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load. J. Clin. Invest. 2018, 128, 4179–4191. [Google Scholar] [CrossRef]
- Herr, A.J.; Ogawa, M.; Lawrence, N.A.; Williams, L.N.; Eggington, J.M.; Singh, M.; Smith, R.A.; Preston, B.D. Mutator suppression and escape from replication error-induced extinction in yeast. PLoS Genet. 2011, 7, e1002282. [Google Scholar] [CrossRef]
- Williams, L.N.; Herr, A.J.; Preston, B.D. Emergence of DNA polymerase epsilon antimutators that escape error-induced extinction in yeast. Genetics 2013, 193, 751–770. [Google Scholar] [CrossRef] [Green Version]
- Albertson, T.M.; Ogawa, M.; Bugni, J.M.; Hays, L.E.; Chen, Y.; Wang, Y.; Treuting, P.M.; Heddle, J.A.; Goldsby, R.E.; Preston, B.D. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17101–17104. [Google Scholar] [CrossRef] [Green Version]
- Barbari, S.R.; Kane, D.P.; Moore, E.A.; Shcherbakova, P.V. Functional analysis of cancer-associated DNA polymerase epsilon variants in Saccharomyces cerevisiae. G3 (Bethesda) 2018, 8, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Zahurancik, W.J.; Klein, S.J.; Suo, Z. Significant contribution of the 3′→5′ exonuclease activity to the high fidelity of nucleotide incorporation catalyzed by human DNA polymerase ϵ. Nucleic Acids Res. 2014, 42, 13853–13860. [Google Scholar] [CrossRef] [Green Version]
- Herzog, M.; Alonso-Perez, E.; Salguero, I.; Warringer, J.; Adams, D.J.; Jackson, S.P.; Puddu, F. Mutagenic mechanisms of cancer-associated DNA polymerase ε alleles. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shinbrot, E.; Henninger, E.E.; Weinhold, N.; Covington, K.R.; Göksenin, A.Y.; Schultz, N.; Chao, H.; Doddapaneni, H.; Muzny, D.M.; Gibbs, R.A.; et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014, 24, 1740–1750. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.W.; Venkatesan, R.N.; Pillaire, M.J.; Hoffmann, J.S.; Sidorova, J.M.; Loeb, L.A. Active site mutations in mammalian DNA polymerase delta alter accuracy and replication fork progression. J. Biol. Chem. 2010, 285, 32264–32272. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Giot, L.; Faye, G. The 3’ to 5’ exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991, 10, 2165–2170. [Google Scholar] [CrossRef]
- Goldsby, R.E.; Hays, L.E.; Chen, X.; Olmsted, E.A.; Slayton, W.B.; Spangrude, G.J.; Preston, B.D. High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading. Proc. Natl. Acad. Sci. USA 2002, 99, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Fazlieva, R.; Spittle, C.S.; Morrissey, D.; Hayashi, H.; Yan, H.; Matsumoto, Y. Proofreading exonuclease activity of human DNA polymerase delta and its effects on lesion-bypass DNA synthesis. Nucleic Acids Res. 2009, 37, 2854–2866. [Google Scholar] [CrossRef] [Green Version]
- Mertz, T.M.; Sharma, S.; Chabes, A.; Shcherbakova, P.V. Colon cancer-associated mutator DNA polymerase delta variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. USA 2015, 112, E2467–E2476. [Google Scholar] [CrossRef] [Green Version]
- Mertz, T.M.; Baranovskiy, A.G.; Wang, J.; Tahirov, T.H.; Shcherbakova, P.V. Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer-associated DNA polymerase δ mutation in human cells. Oncogene 2017, 36, 4427–4433. [Google Scholar] [CrossRef] [Green Version]
- Bellelli, R.; Borel, V.; Logan, C.; Svendsen, J.; Cox, D.E.; Nye, E.; Metcalfe, K.; O’Connell, S.M.; Stamp, G.; Flynn, H.R.; et al. Polε instability drives replication stress, abnormal development, and tumorigenesis. Mol. Cell 2018, 70, 707–721. [Google Scholar] [CrossRef]
- Wittschieben, J.P.; Reshmi, S.C.; Gollin, S.M.; Wood, R.D. Loss of DNA polymerase z causes chromosomal instability in mammalian cells. Cancer Res. 2006, 66, 134–142. [Google Scholar] [CrossRef] [Green Version]

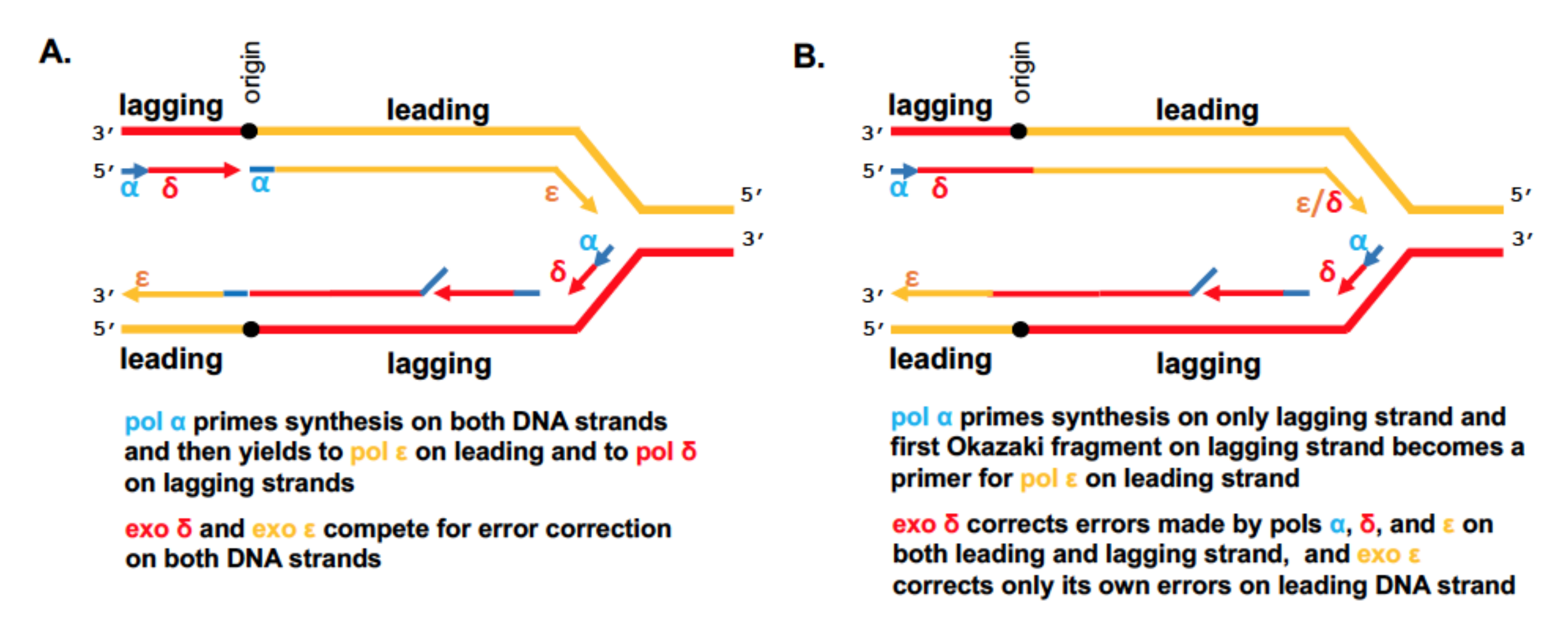
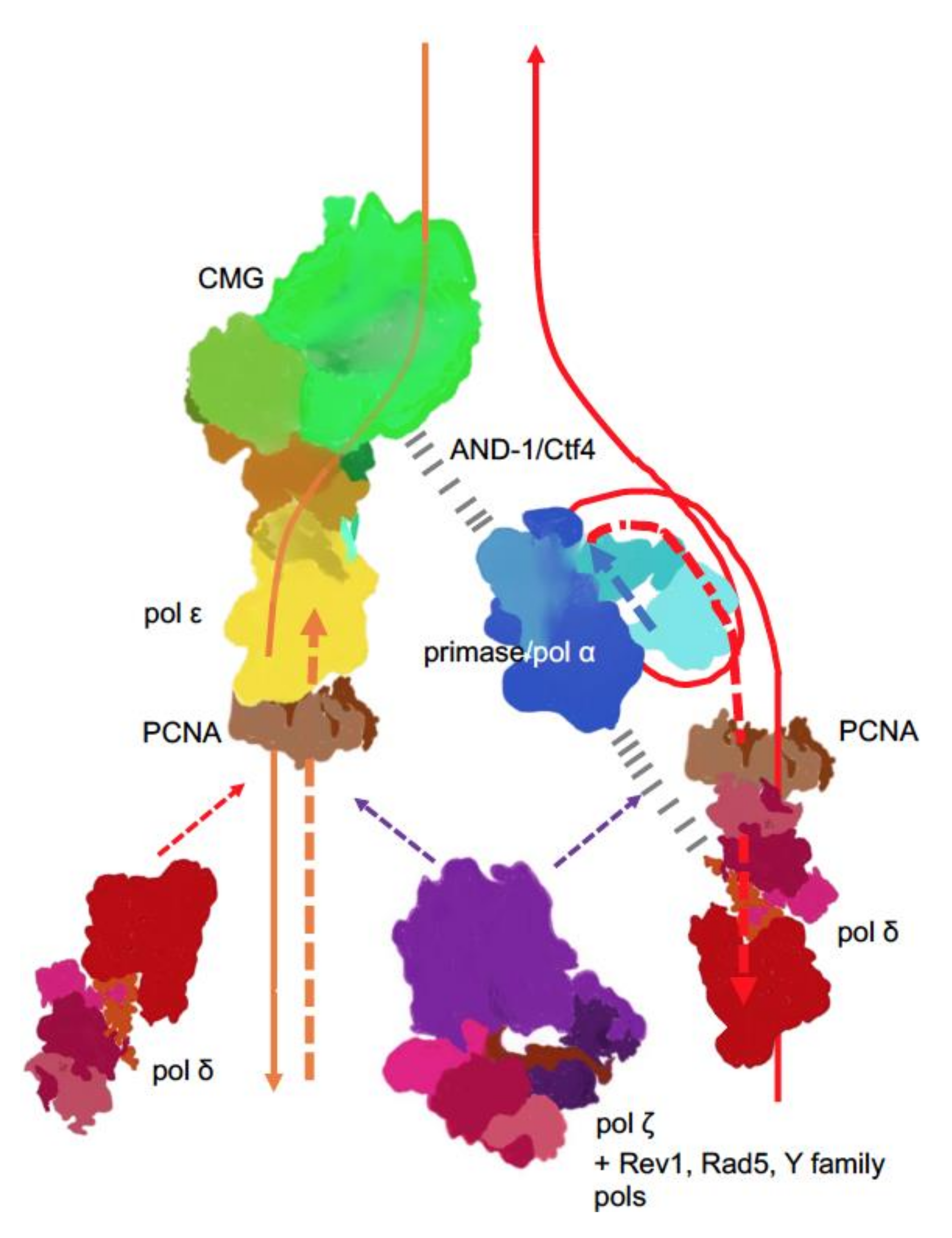
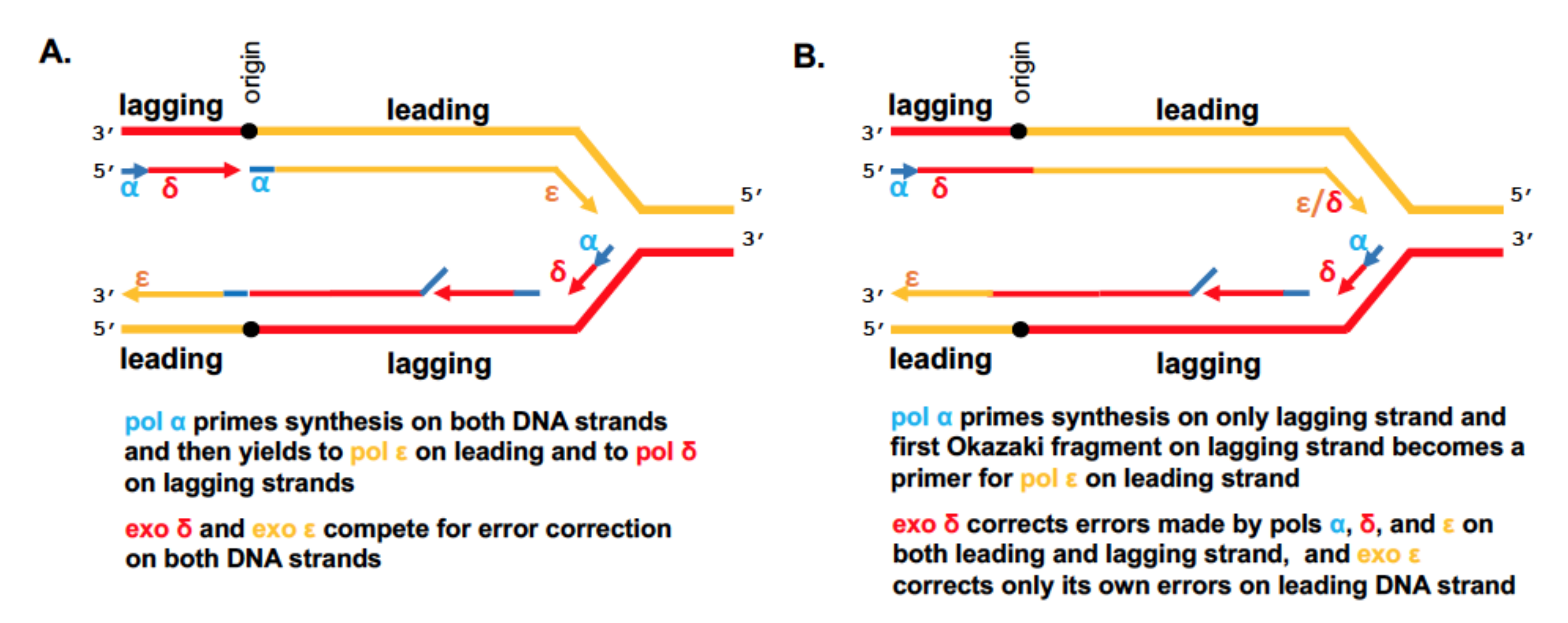
 ) is present in each of the four pol complexes.
) is present in each of the four pol complexes.
) is present in each of the four pol complexes.
) is present in each of the four pol complexes.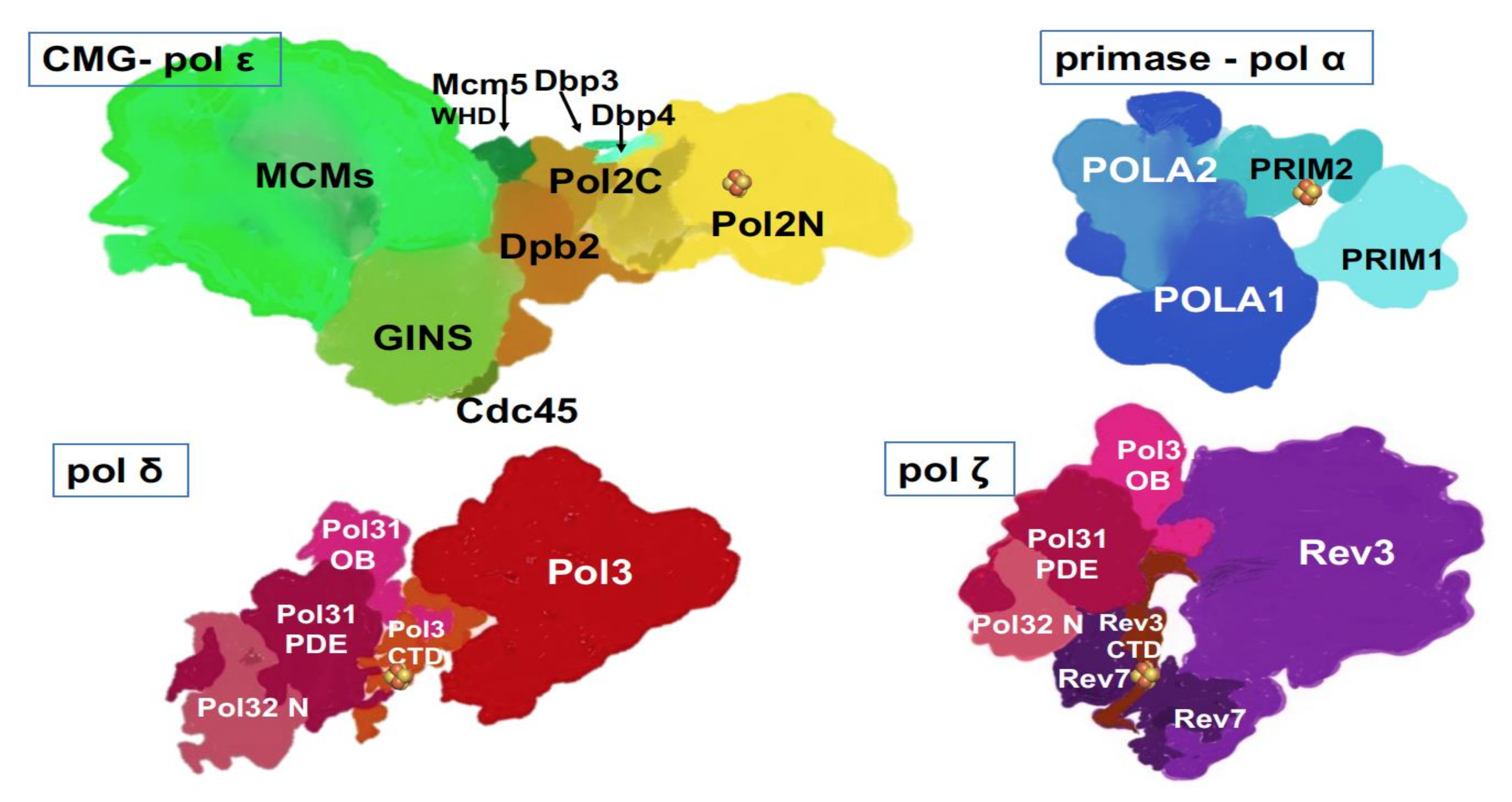

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymerase | Subunit ** | Yeast, S. cerevisiae | Human, H. sapiens | ||||
|---|---|---|---|---|---|---|---|
| Gene | Protein, (Size, kDa) | Also Known As: | Gene | Protein, (Size, kDa) | Also Known As: | ||
| primase -pol α | Small (catalytic primase) | PRI1 | Pri1 (48) | YIR008C | PRIM1 | PRIM1 (50) | p48, p49 |
| Large of primase | PRI2 | Pri2 (62) | YKL045W | PRIM2 | PRIM2 (59) | PRIM2A, p58 | |
| Catalytic | POL1 | Pol1 (167) | CDC17, CRT5, HPR3, YNL102W | POLA1 | POLA1 (166) | POLA, NSX, p180 | |
| B-subunit | POL12 | Pol12 (79) | YBL035C | POLA2 | POLA2 (66) | FLJ21662, p70 | |
| pol ε | Catalytic | POL2 | Pol2 (256) | DUN2
, YNL262W | POLE | POLE (262) | FILS, POLE1, IMADEI, CRCS12, p261 |
| B-subunit | DPB2 | Dpb2 (78) | YPR175W | POLE2 | POLE2 (60) | DPE2, p59 | |
| Third | DPB3 | Dpb3 (23) | YBR278W | POLE3 | POLE3 (17) | CHRAC2, YBL1, p17 | |
| Fourth | DPB4 | Dpb4 (22) | YDR121W | POLE4 | POLE4 (12) | YHHQ1, p12 | |
| pol δ | Catalytic | POL3 | Pol3 (125) | CDC2, HPR2, TEX1
, YDL102W | POLD1 | POLD1 (124) | CDC2, MDPL, CRCS10, p125 |
| B-subunit | POL31 | Pol31 (55) | HYS2, HUS2, SDP5, YJR006W | POLD2 | POLD2 (51) | p50 | |
| Third | POL32 | Pol32 (40) | REV5, YJR043C | POLD3 | POLD3 (51) | PPP1R128, KIAA0039, p66 | |
| Fourth | - | - | POLD4 | POLD4 (12) | POLDS, p12 | ||
| pol ζ | Catalytic | REV3 | Rev3 (173) | PSO1
, YPL167C | REV3L | REV3L (353) | REV3, HREV3, POLZ p353 |
| Second | REV7 | Rev7 (29) | YIL139C | MAD2L2 | MAD2L2 (24) | hREV7, p30, FANCV, MAD2B, POLZ2 | |
| B-subunit | POL31 | Pol31 (55) | HYS2, HUS2, SDP5 | POLD2 | POLD2 (51) | p50 | |
| Fourth | POL32 | Pol32 (40) | REV5 | POLD3 | POLD3 (51) | PPP1R128, p66 | |
| Pol Amino Acid Change; (Exo Activity) § | Pol Region, Sequence, Conservation. Changed Amino Acids Are Underlined. Amino Acids Different from Human Protein Are Highlighted Grey. | Recurrently Found in Cancers *; Predominant Types of Cancer [124,139] and cBioPortal | Mutation Burden in Genomes of Tumors with the Change ** | Sporadic (s) or Hereditary (h) | Yeast Variants: | Mice Variants: | ||
|---|---|---|---|---|---|---|---|---|
| Allele Name; Amino Acid Change; (Exo Activity) | Mutator Effect Relative to Wild-Type; (Method of Determination) | Allele Variant (Exo Activity) | Mutator Effects Relative to Wild-Type; (Method of Determination); Cancer Incidence; Predominant Types of Cancer | |||||
| POLE | ||||||||
| D275A; E277A; (-) [122,140] | Exo IHs PVVLAFDIETTKLPMm PVVLAFDIETTKLPSc PVVMAFDIETTKPP | not found, classic model exo− variant | n/a | n/a | pol2–4; D290A;E292A; (-) [125] | 5; (Canr) [20] 2.9; (Canr) [126] 4; (SNVs /genome) [141] ~11; (Lyp-) [141] | PoleD272A;E274A/D272,E274A(-) *** | 10; (derivatives of BigBlueTM mice); [138] >70; (ouabain resistance) or 170; thioguanine resistance in MEFs) [138] ;medium; intestine adenocarcinoma, nodal lymphoma [138] |
| D275V; (-) ***D275A; (-) | Exo IHs PVVLAFDIETTKLPMm PVVLAFDIETTKLPSc PVVMAFDIETTKPP | +; endometrial, breast, glioblastoma, colorectal, lung | med | s | pol2-D290V; (-) | 2.3; (Canr) [133] ~9; (Lyp-) [141] | ||
| P286R; (−/+) [122] | Exo IHs FDIETTKLPLKFPDMm FDIETTKLPLKFPDSc FDIETTKPPLKFPD | +++; colon, endometrial, ovarian, high grade glyoma pancreatic, breast, prostate, bladder and other cancers | ultra-high | s | P301R; (-/+) [125] | 150; (Canr) [126]~200; (Lyp-) [141] 63; (SNVs/ genome) [141] | PoleP286R/+ (P286R; nd | ~40; (NGS #) [135] >100; (NGS) [122]; high; thymic lymphomas, lung adenocarcinomas, angiosarcoma [135]; thymic lymphomas, splenic lymphomas [122] |
| P286H; (−/+) [122,142] P286L | Same region, but different amino acid change | +; colon, glioblastoma, stomach | ultra-high | s | P301H; nd | 13; (Canr) [133] | ||
| S459F; (-) [122,142] | ExoIIIHs TYSVSDAVATYYMm TYSVSDAVATYYSc EYSVSDAVATYY | ++; colon, endometrial, glioblastoma, duodenal | high | s | S474F; nd | 30; (Canr) [133] | PoleS459F/S459F (S459F); nd | >220; (NGS); high; thymic/splenic lymphomas [122] |
| F367S; (+/−) [122,142] | Exo IIHs MVTYNGDFFDWPFMm MVTYNGDFFDWPFSc ISTFNGDFFDWPF | +; endometrial, colon | ultra-high | s | F382S; nd | 17; (Canr) [133] | ||
| P436R; nd | Exo VHs AKLGYDPVELDPMm AKLGYDPVELDPSc AKLGYNPIELDP | +; endometrial, colorectal | ultra-high | s | P451R; nd | 5.2; (Canr) [133] | ||
| V411L; (+/-) [142] | Hs CLRWVKRDSYLPVMm CLRWVKRDSYLPVSc CFRWVKRDSYLPQ | +++; +; endometrial, colorectal, glioblastoma, kidney cancer, ovarian medulloblastoma, urinary tract, cervix, stomach | ultra-high | s h | V426L; nd | 1.2; (Canr) [133] ~5; (Lyp-) [141] 1.8; (SNVs/ genome) [141] | ||
| L424V; (+/-) [122,142] | Exo IVHs LPVGSHNLKAAAKMm LPVGSHNLKAAAKSc LPQGSQGLKAVTQ | +; +++; colorectal, endometrial, lung, breast, glyoblastoma, duodenal [122] | med | s h | L439V; nd | 5.2; (Canr) [133] ~33; (Lyp-) [141] 7; (SNVs/ genome) [141] | ||
| POLD1 | ||||||||
| D316A; E318A; (-) | Exo IHs LRVLSFDIECAGRKMm LRVLSFDIECAGRKSc LRIMSFDIECAGRI | Double change not found, but +; D316N or D316G; endometrial | high | s | pol3–01; D321A;E323A; (+) [22] | 130; (FOAr) [21] 110; (Canr) [22] | ||
| D402A; (-) [143] | ExoIIHs TGYNIQNFDLPYLIMm TGYNIQNFDLPYLISc TGYNTTNFDIPYLL | not found in cBio, but other changes of the nearby amino acids in motif +; breast adeno carcinoma, melanoma | Q399H med P404Shigh | s | pol3–4DA; D407A; (-) [144] pol3–4DV; D407V;nd | 55; (Canr) [22] 76; (Canr) [22] | Pold1D400A/D400A (-)[145] | 3; (derivatives of BigBlueTM mice) [138] ;>10; (ouabain resistance in MEFs) [145] 6; (fibrosarcoma cell line) [145] >50; (ouabain or thioguanine resistance in MEFs) [138] ;high; thymic lymphomas, tail skin carcinoma, lung adenocarcinoma |
| D515A; (-) [146] | Exo IIIHs AVYCLKDAYLPLRLMm AVYCLKDAFLPLRLSc AVYCLKDAYLPLRL | +; currently not found, but D515N variant was detected in melanoma | med | s | pol3–5DV; D520V; (-) [22] | 20; (Canr) [22] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlov, Y.I.; Zhuk, A.S.; Stepchenkova, E.I. DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer. Cancers 2020, 12, 3489. https://doi.org/10.3390/cancers12123489
Pavlov YI, Zhuk AS, Stepchenkova EI. DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer. Cancers. 2020; 12(12):3489. https://doi.org/10.3390/cancers12123489
Chicago/Turabian StylePavlov, Youri I., Anna S. Zhuk, and Elena I. Stepchenkova. 2020. "DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer" Cancers 12, no. 12: 3489. https://doi.org/10.3390/cancers12123489
APA StylePavlov, Y. I., Zhuk, A. S., & Stepchenkova, E. I. (2020). DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer. Cancers, 12(12), 3489. https://doi.org/10.3390/cancers12123489