Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
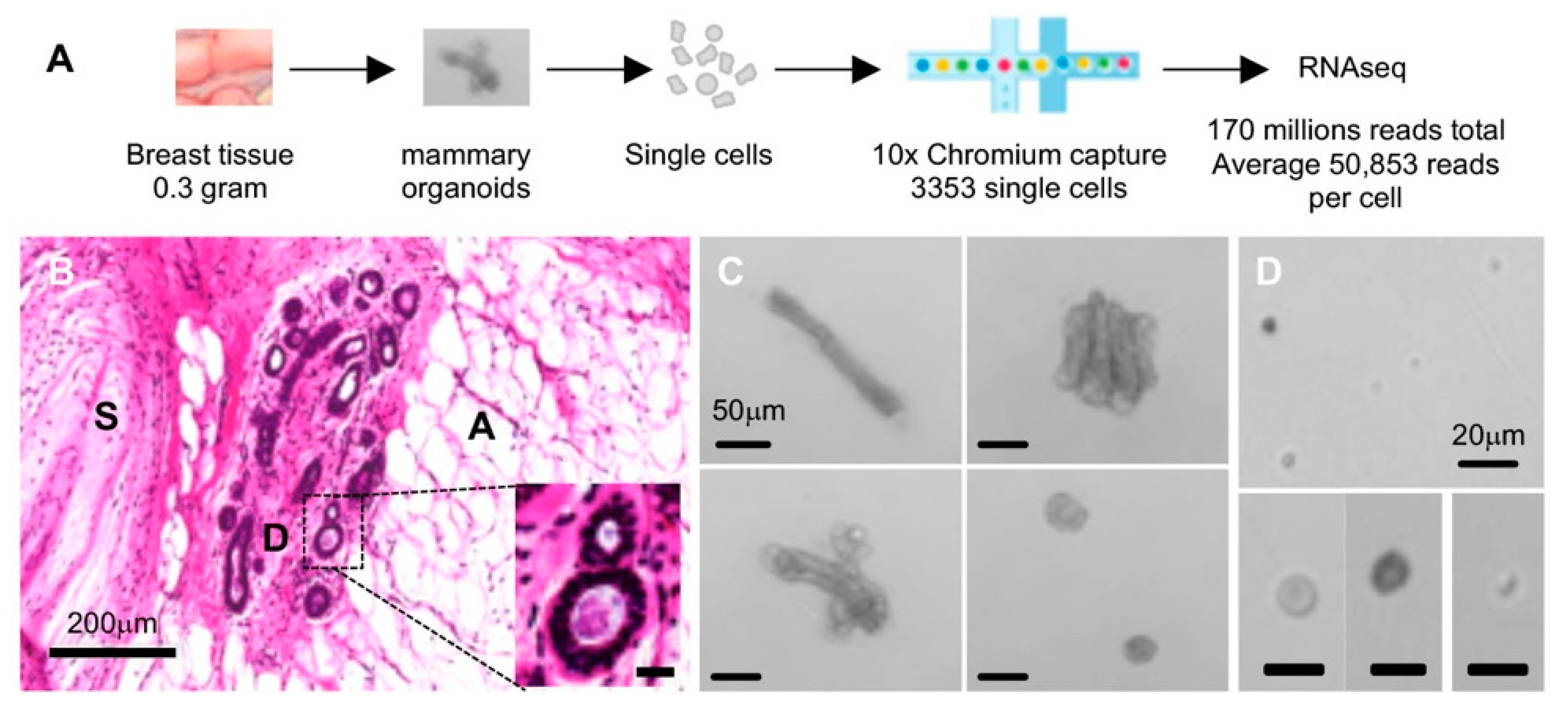
2.1. Single-Cell Isolation and Single-Cell RNA Sequencing (scRNAseq) of Normal Breast Cells
2.2. Single-Cell Transcriptomic Landscape of the Breast Shows 10 Distinct Cell Clusters
2.3. EpCAM and ITGA6 Expression in Breast Single-Cell Clusters
2.4. Cytokeratin Gene Expression in the Three Breast Epithelial Cell Types
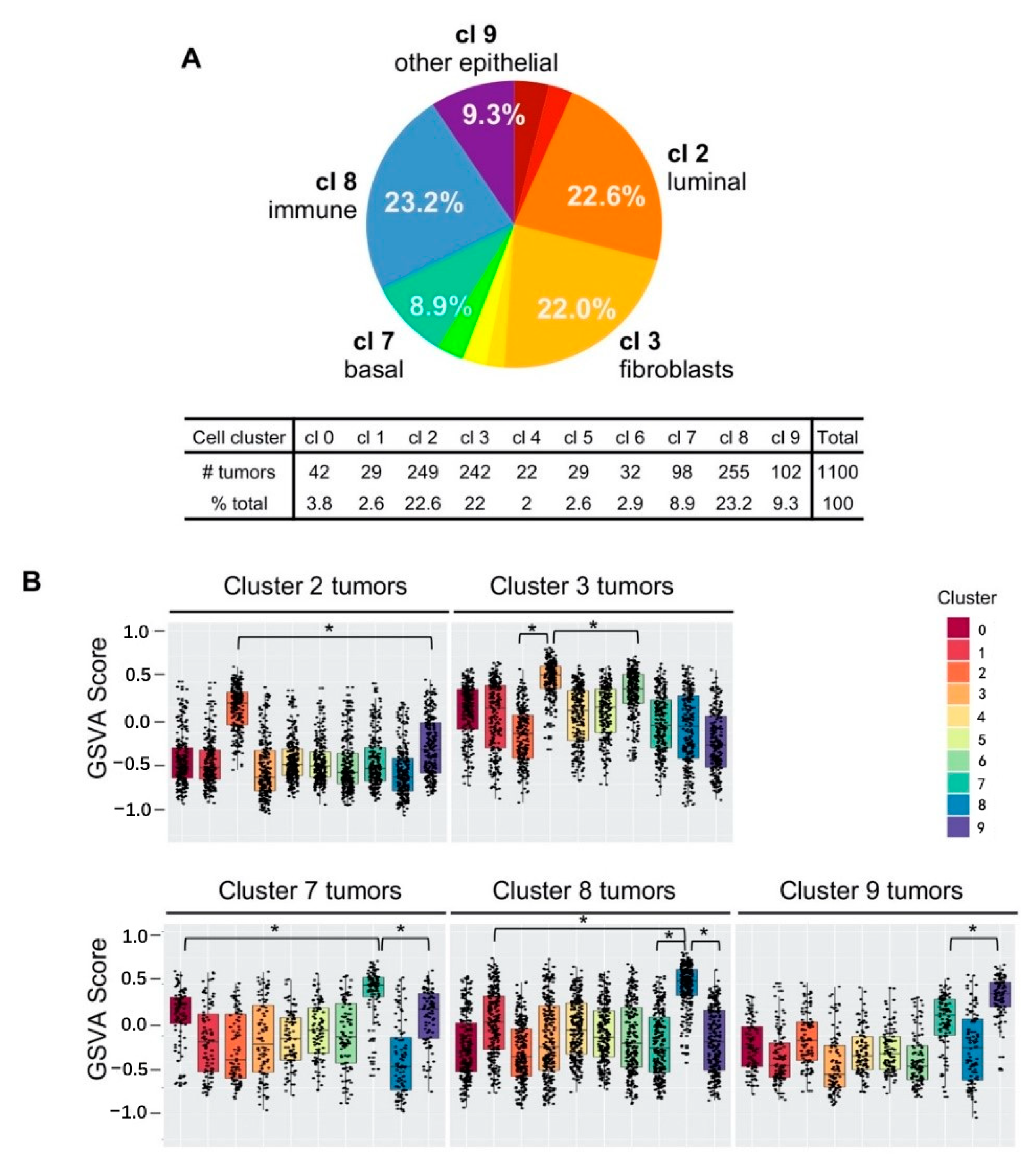
2.5. Five Cell Cluster Signatures Representing 86% of Breast Tumors in TCGA Dataset
2.6. Single-Cell Cluster Signatures Correlating with the Breast Cancer Subtypes
2.7. Poor Patient Survival in Cluster 9 TNBC Tumors
3. Discussions
3.1. Inclusive Single-Cell Landscape of the Normal Breast
3.2. Three Epithelial Cell Types (Clusterd 2, 7, and 9)
3.3. Nonepithelial Cells in the Breast That Do Not Contribute to Breast Cancer
3.4. Fibroblasts (Cluster 3 and 6) and Cancer-Associated Fibroblasts (CAFs)
3.5. Breast Cancer Subtypes Related to the Normal Cell Types in the Breast
4. Methods and Materials
4.1. Breast Tissue Sample and Tissue Processing
4.2. Single-Cell Isolation and Sequencing
4.3. Single-Cell RNA Sequencing Data Analysis
4.4. The Cancer Genome Atlas (TCGA) Data Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daniel, C.W.; Smith, G.H. The mammary gland: A model for development. J. Mammary Gland Biol. Neoplasia 1999, 4, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wellings, S.R.; Jensen, H.M. On the Origin and Progression of Ductal Carcinoma in the Human Breast2. J. Natl. Cancer Inst. 1973, 50, 1111–1118. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Wetzels, R.; Ramaekers, F. Intermediate filament protein expression in normal and malignant human mammary epithelial cells. Cancer Treat. Res. 1992, 61, 355–378. [Google Scholar]
- Gusterson, B.A.; Ross, D.T.; Heath, V.J.; Stein, T. Basal cytokeratins and their relationship to the cellular origin and functional classification of breast cancer. Breast Cancer Res. 2005, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullenix, P.S.; Brown, T.A.; Meyers, M.O.; Giles, L.R.; Sigurdson, E.R.; Boraas, M.C.; Hoffman, J.P.; Eisenberg, B.L.; Torosian, M.H. The association of cytokeratin-only-positive sentinel lymph nodes and subsequent metastases in breast cancer. Am. J. Surg. 2005, 189, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Lemaire, C.A.; Triboulet, M.; Casey, K.M.; Heirich, K.; Renier, C.; Vilches-Moure, J.G.; Gupta, R.; Razmara, A.M.; Zhang, H.; et al. Investigating circulating tumor cells and distant metastases in patient-derived orthotopic xenograft models of triple-negative breast cancer. Breast Cancer Res. 2019, 21, 1–16. [Google Scholar] [CrossRef]
- Heath, J.R.; Ribas, A.; Mischel, P.S. Single-cell analysis tools for drug discovery and development. Nat. Rev. Drug Discov. 2016, 15, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Kim, C.; Sei, E.; Foukakis, T.; Crosetto, N.; Chan, L.-K.; Srinivasan, M.; Zhang, H.; Meric-Bernstam, F.; Navin, N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.; Fab, K.C.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.; Ward, T.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Pervolarakis, N.; Blake, K.; Ma, D.; Davis, R.T.; James, N.; Phung, A.T.; Willey, E.; Kumar, R.; Jabart, E.; et al. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Mammary development meets cancer genomics. Nat. Med. 2009, 15, 842–844. [Google Scholar] [CrossRef]
- Cristea, S.; Polyak, K. Dissecting the mammary gland one cell at a time. Nat. Commun. 2018, 9, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Stingl, J.; Raouf, A.; Emerman, J.T.; Eaves, C.J.; Stingl, J.C. Epithelial Progenitors in the Normal Human Mammary Gland. J. Mammary Gland Biol. Neoplasia 2005, 10, 49–59. [Google Scholar] [CrossRef]
- Visvader, J.; Stingl, J. Mammary stem cells and the differentiation hierarchy: Current status and perspectives. Genes Dev. 2014, 28, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Welm, A.L.; Kim, S.; Welm, B.E.; Bishop, J.M. MET and MYC cooperate in mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 4324–4329. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 1–3. [Google Scholar] [CrossRef]
- Hendricks, W.P.D.; Briones, N.; Halperin, R.F.; Facista, S.; Heaton, P.R.; Mahadevan, D.; Kim, S. PD-1-Associated Gene Expression Signature of Neoadjuvant Trastuzumab-Treated Tumors Correlates with Patient Survival in HER2-Positive Breast Cancer. Cancers 2019, 11, 1566. [Google Scholar] [CrossRef] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehadeh, L.A.; Webster, K.A.; Hare, J.M.; Vazquez-Padron, R.I. Dynamic Regulation of Vascular Myosin Light Chain (MYL9) with Injury and Aging. PLoS ONE 2011, 6, e25855. [Google Scholar] [CrossRef] [PubMed]
- Crnkovic, S.; Marsh, L.M.; El Agha, E.; Voswinckel, R.; Ghanim, B.; Klepetko, W.; Stacher-Priehse, E.; Olschewski, H.; Bloch, W.; Bellusci, S.; et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J. Pathol. 2018, 244, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Fan, Y.; Wan, J. Screening of key genes of unruptured intracranial aneurysms by using DNA microarray data analysis techniques. Genet. Mol. Res. 2014, 13, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.F.; Heng, J.S.; Luo, C.; Castanon, R.G.; Nery, J.R.; Rattner, A.; Goff, L.A.; Ecker, J.R.; Nathans, J. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, T.; Adriance, M.C.; Sternlicht, M.D.; Petersen, O.W.; Bissell, M.J. Myoepithelial Cells: Their Origin and Function in Breast Morphogenesis and Neoplasia. J. Mammary Gland Biol. Neoplasia 2006, 10, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assinder, S.; Stanton, J.-A.L.; Prasad, P.D. Transgelin: An actin-binding protein and tumour suppressor. Int. J. Biochem. Cell Biol. 2009, 41, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Inman, J.L.; Robertson, C.; Mott, J.D.; Bissell, M.J. Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development 2015, 142, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2010, 15, 635–646. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef]
- Nguyen, A.; Khoo, W.H.; Moran, I.; Croucher, P.I.; Phan, T.G. Single Cell RNA Sequencing of Rare Immune Cell Populations. Front. Immunol. 2018, 9, 1553. [Google Scholar] [CrossRef] [PubMed]
- Alford, D.; Taylor-Papadimitriou, J. Cell adhesion molecules in the normal and cancerous mammary gland. J. Mammary Gland Biol. Neoplasia 1996, 1, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Moll, R.; Divo, M.; Langbein, L. The human keratins: Biology and pathology. Histochem. Cell Biol. 2008, 129, 705–733. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finak, G.; Bertos, N.R.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Wennmalm, K.; Ostman, A.; Bergh, J. Stromal signature identifies basal breast cancers. Nat. Med. 2009, 15, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Carey, L.A.; Adamo, B.; Vidal, M.; Tabernero, J.; Cortés, J.; Parker, J.S.; Perou, C.M.; Baselga, J. Molecular Features and Survival Outcomes of the Intrinsic Subtypes Within HER2-Positive Breast Cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-Infiltrating CD8+ Lymphocytes Predict Clinical Outcome in Breast Cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Ibrahim, E.M.; Al-Foheidi, M.E.; Al-Mansour, M.M.; Kazkaz, G.A. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2014, 148, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Papalexi, E.; Satija, R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.-C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pal, B.; Chen, Y.; Vaillant, F.; Jamieson, P.; Gordon, L.; Rios, A.C.; Wilcox, S.; Fu, N.; Liu, K.H.; Jackling, F.C.; et al. Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Soysal, S.D.; Tzankov, A.; Muenst, S. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Raouf, A.; Zhao, Y.; To, K.; Stingl, J.; Delaney, A.; Barbara, M.; Iscove, N.; Jones, S.; McKinney, S.; Emerman, J.; et al. Transcriptome Analysis of the Normal Human Mammary Cell Commitment and Differentiation Process. Cell Stem Cell 2008, 3, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Morabito, S.J.; Kessenbrock, K.; Enver, T.; Meyer, K.B.; Teschendorff, A.E. Single-cell landscape in mammary epithelium reveals bipotent-like cells associated with breast cancer risk and outcome. Commun. Biol. 2019, 2, 1–13. [Google Scholar] [CrossRef]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nat. Cell Biol. 2014, 506, 322–327. [Google Scholar] [CrossRef]
- Anderson, W.F.; Chatterjee, N.; Ershler, W.B.; Brawley, O.W. Estrogen Receptor Breast Cancer Phenotypes in the Surveillance, Epidemiology, and End Results Database. Breast Cancer Res. Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef]
- Barton, M.; Yanagisawa, M. Endothelin: 30 Years From Discovery to Therapy. Hypertension 2019, 74, 1232–1265. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, B.M.; Weinstein, B.M. Arterial—Venous Specification During Development. Circ. Res. 2009, 104, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, R.J.; Oh, S.Y. Breast Cancer-Associated Fibroblasts: Where We Are and Where We Need to Go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehner, C.; Radisky, D.C. Triggering the landslide: The tumor-promotional effects of myofibroblasts. Exp. Cell Res. 2013, 319, 1657–1662. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24, S26–S35. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Cluster | Number of Cells | % Total Cells | Genes | Proposed Cell Type |
|---|---|---|---|---|
| 0 | 769 | 24.1 | ACTA2, MYH11, MYL9, MYLK, TAGLN | Smooth muscle |
| 1 | 749 | 23.5 | EMCN, ENG, PLVAP, SELE, SELP | Endothelial |
| 2 | 453 | 14.2 | * KRT18, KRT19, KRT7, KRT8 | Luminal epithelial |
| 3 | 336 | 10.5 | COL1A1/A2, COL3A1, COL6A1/A2/A3, ALDH1A1 | Stroma fibroblasts |
| 4 | 305 | 9.6 | EDN1, EFNB2, ELTD1, ESAM | Endothelial subset |
| 5 | 188 | 5.9 | Adipocyte markers: APOE, CFD, STEAP4 Macrophage markers: CCL2, CEBPD Fibroblast markers: COL4A1, COL6A3 EMT markers: SNAI2, TGFB1 | Mesenchymal progenitor |
| 6 | 161 | 5.0 | COL14A1, COL1A1/A2, COL6A2, ALDH1A1 | Stroma fibroblast subset |
| 7 | 153 | 4.8 | * KRT5, KRT14, KRT17, ACTA2, TAGLN | Basal/myoepithelial |
| 8 | 48 | 1.5 | C2, CD3D, CD7, CD8A, CD69, IL1B, TNF, GZMK | Immune CTL |
| 9 | 31 | 1.0 | EPCAM, * KRT6B, KRT15, KRT16, KRT81, KRT23 | Other epithelial |
| All | 3193 | 100.0% |
| Cluster | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| EPCAM | 0.01 ± 0.09 | 0 | 0.42 ± 0.59 | 0.01 ± 0.07 | 0.01 ± 0.12 | 0 ± 0.05 | 0 | 0.12 ± 0.38 | 0 | 1.15 ± 0.68 |
| ITGA6 | 0.01 ± 0.08 | 0.59 ± 0.72 | 0.01 ± 0.11 | 0.02 ± 0.17 | 0.35 ± 0.62 | 0.02 ± 0.17 | 0 | 0.22 ± 0.51 | 0.02 ± 0.12 | 0.15 ± 0.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, S.; Hebert, L.L.; Eschbacher, J.M.; Kim, S. Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer. Cancers 2020, 12, 3639. https://doi.org/10.3390/cancers12123639
Peng S, Hebert LL, Eschbacher JM, Kim S. Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer. Cancers. 2020; 12(12):3639. https://doi.org/10.3390/cancers12123639
Chicago/Turabian StylePeng, Sen, Lora L. Hebert, Jennifer M. Eschbacher, and Suwon Kim. 2020. "Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer" Cancers 12, no. 12: 3639. https://doi.org/10.3390/cancers12123639
APA StylePeng, S., Hebert, L. L., Eschbacher, J. M., & Kim, S. (2020). Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer. Cancers, 12(12), 3639. https://doi.org/10.3390/cancers12123639