Single Cell Detection of the p53 Protein by Mass Cytometry

 and
and
Abstract
:Simple Summary
Abstract
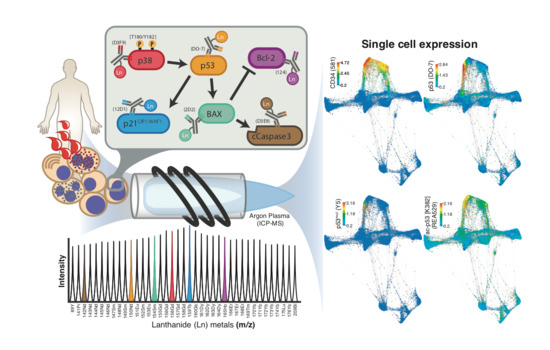
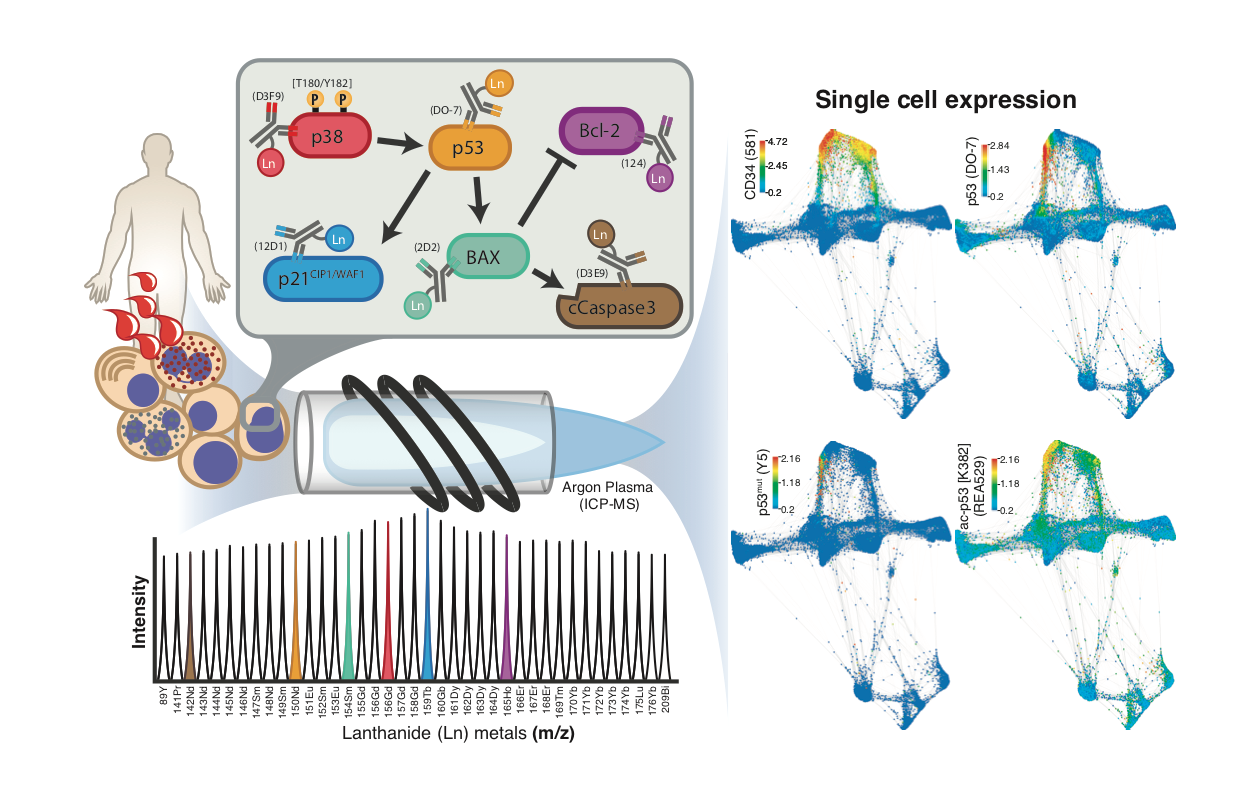
1. Introduction
2. Results
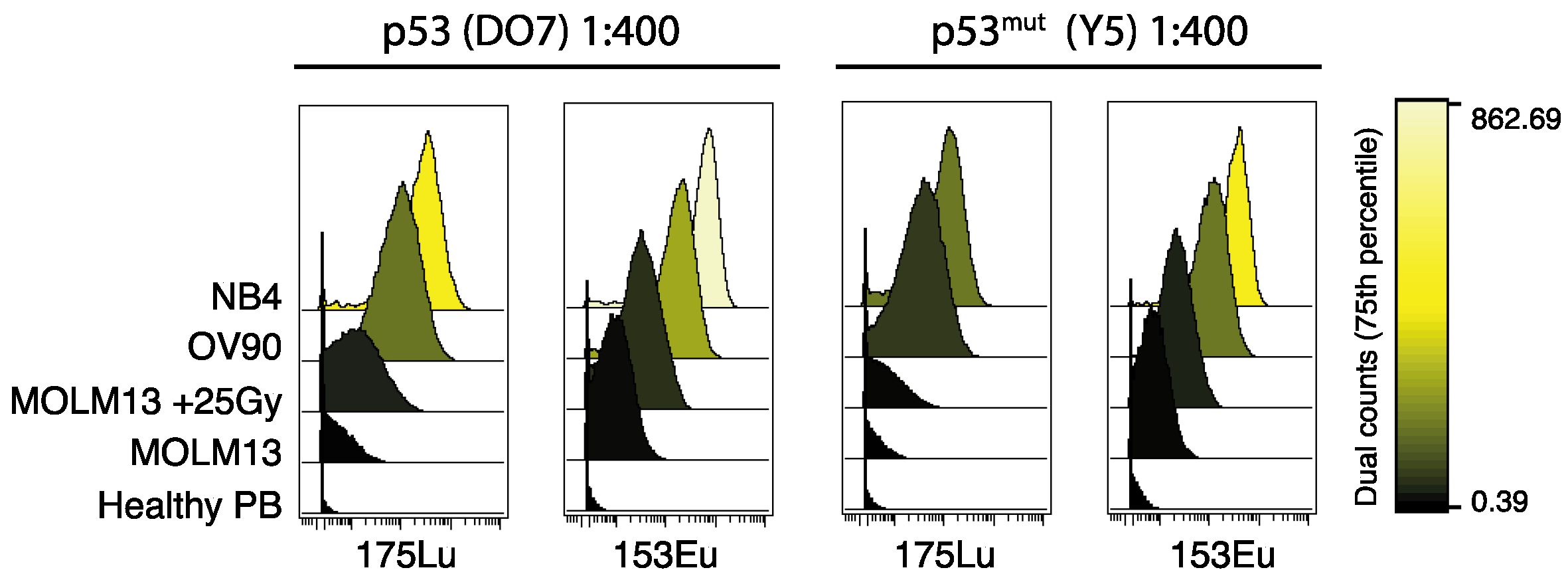
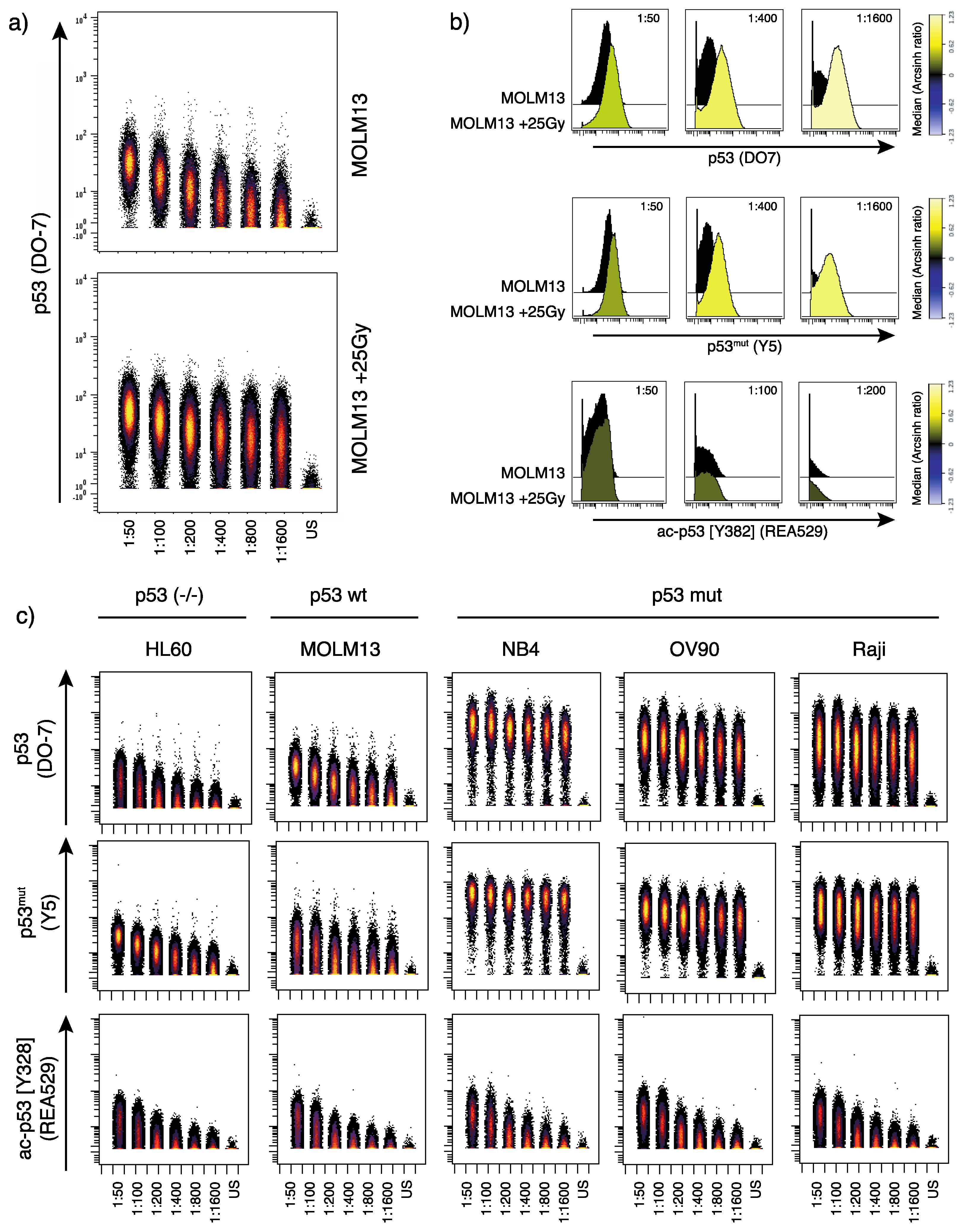
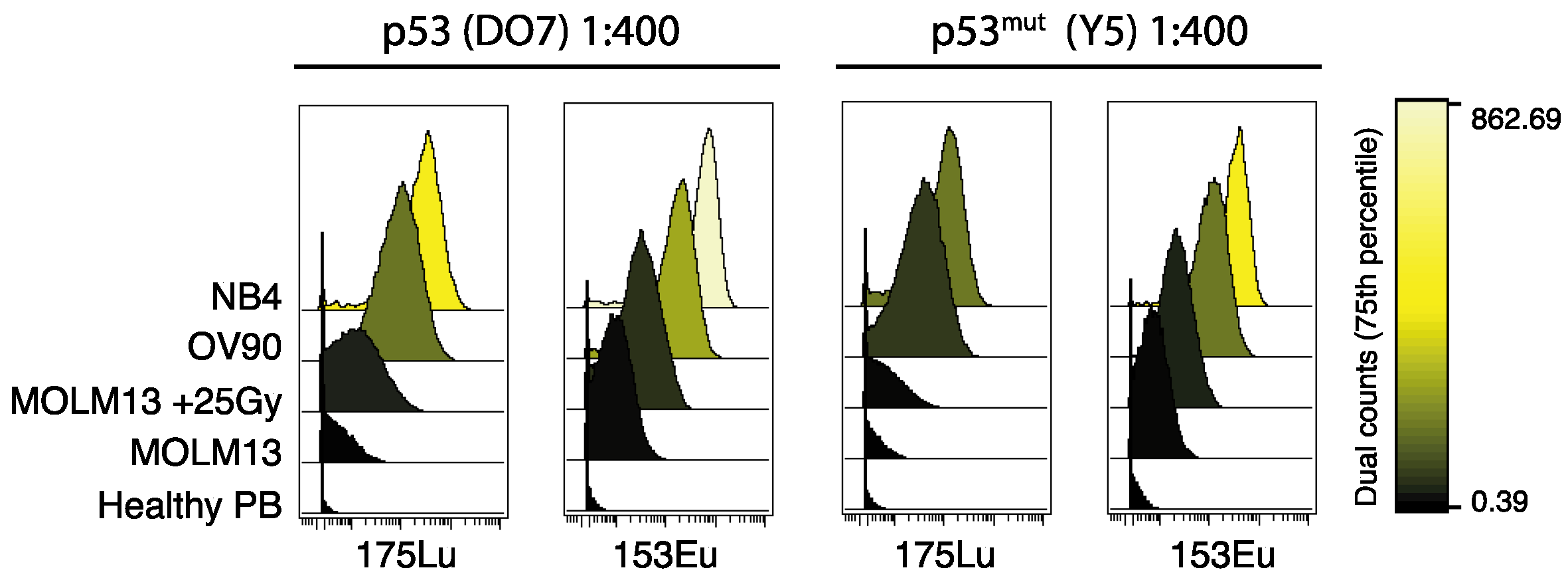
2.1. Titration and Validation of p53-Specific Antibodies for Mass Cytometry
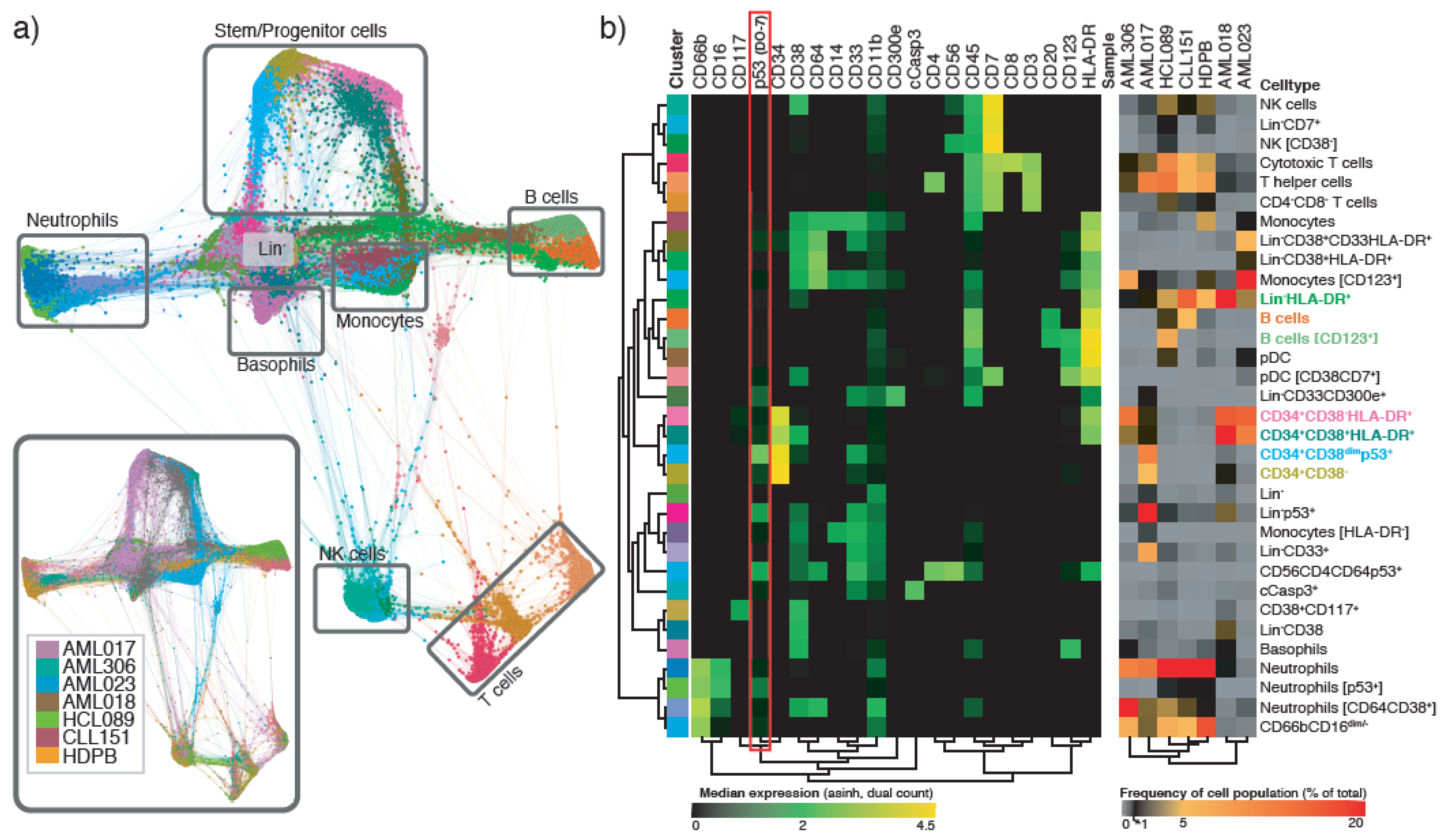
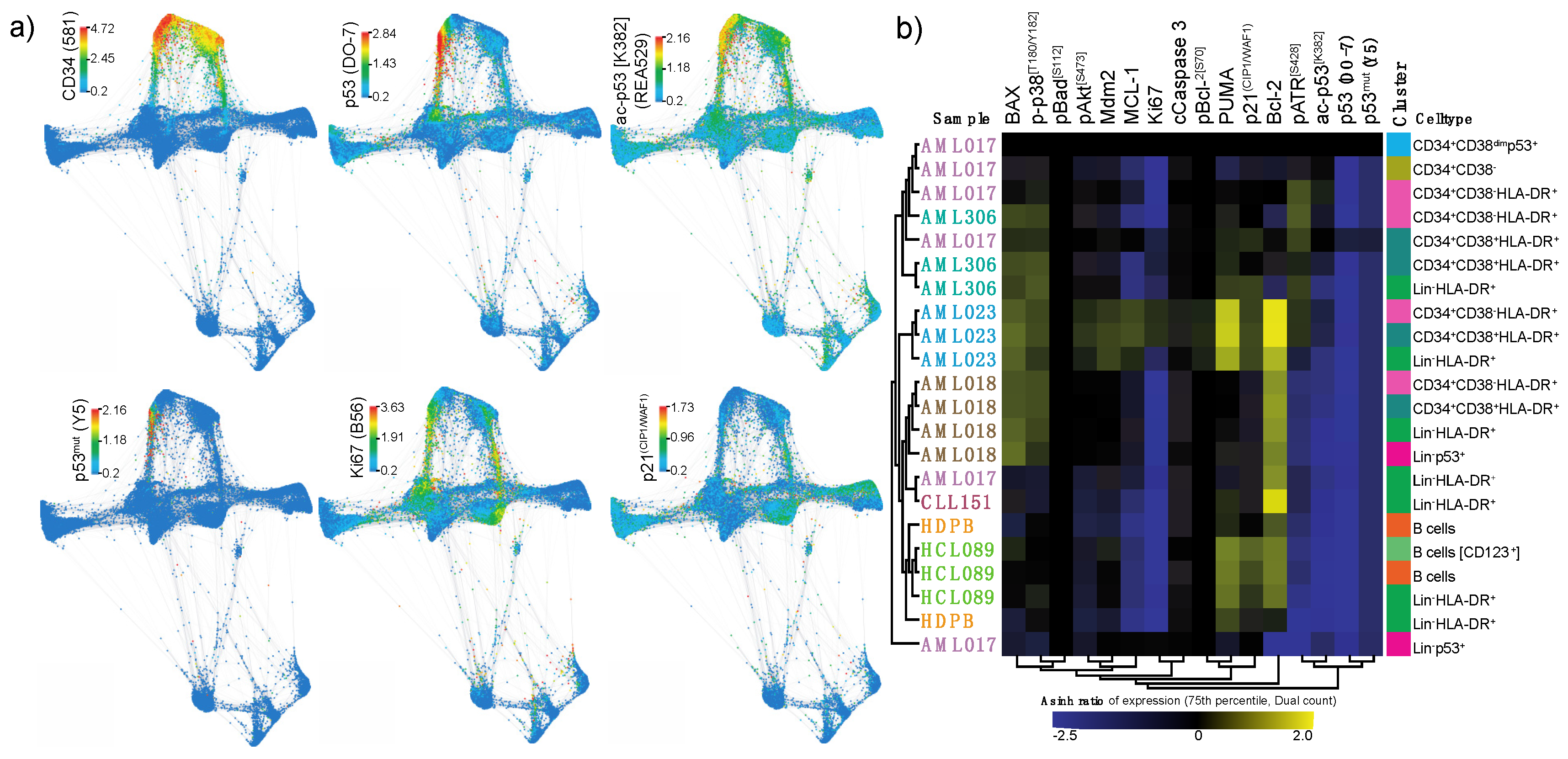
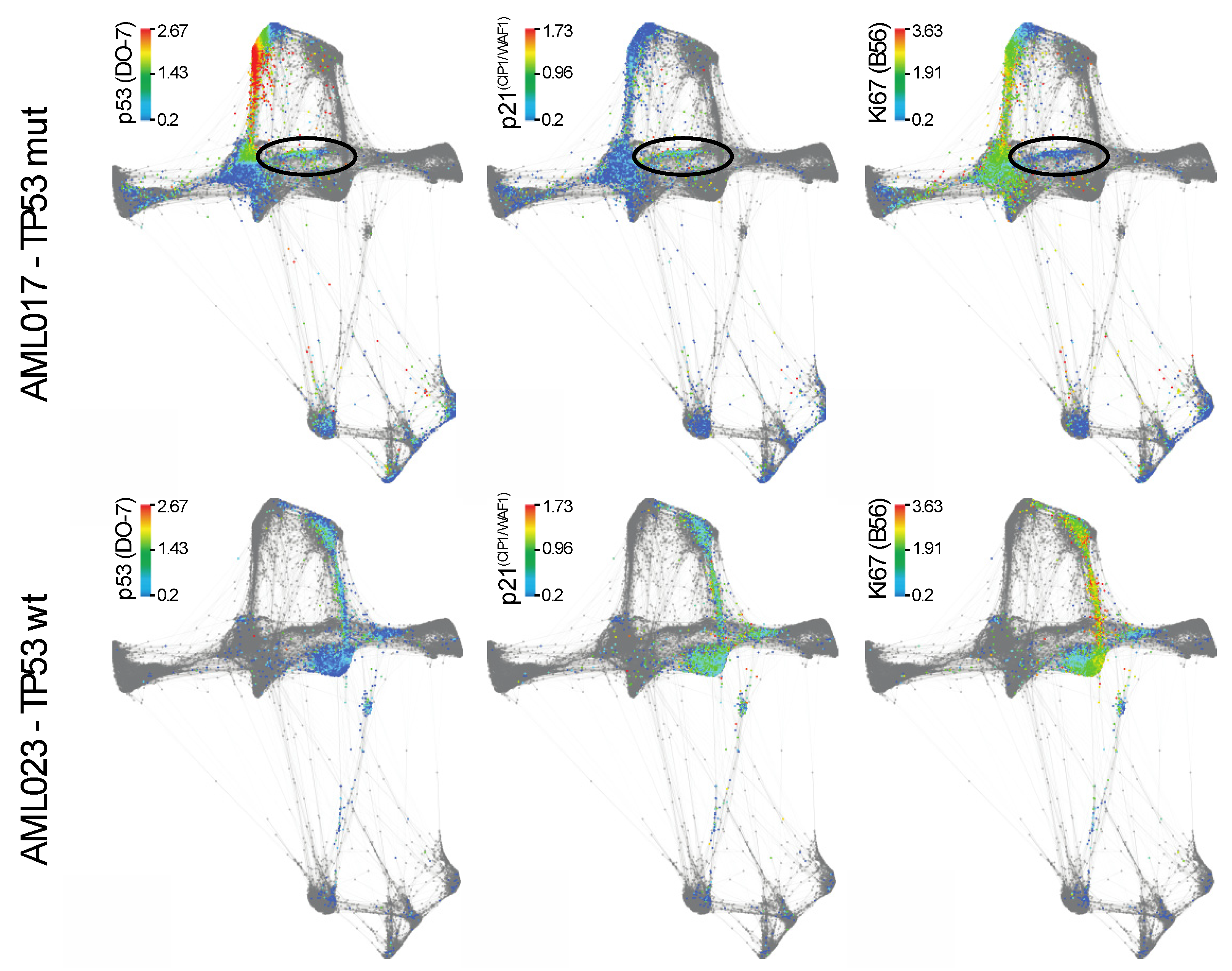
2.2. The Potential of Mass Cytometry for Detection of p53 and p53-Related Proteins in Primary Samples
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.D.; Chen, P.; Levine, A.J. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054. [Google Scholar] [CrossRef] [Green Version]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Short, N.J.; Rytting, M.E.; Cortes, J.E. Acute myeloid leukaemia. Lancet 2018, 392, 593–606. [Google Scholar] [CrossRef]
- Ånensen, N.; Hjelle, S.M.; Van Belle, W.; Haaland, I.; Silden, E.; Bourdon, J.-C.; Hovland, R.; Taskén, K.; Knappskog, S.; Lønning, P.E.; et al. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene 2012, 31, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001, 268, 2764–2772. [Google Scholar] [CrossRef]
- Meek, D.W. Regulation of the p53 response and its relationship to cancer1. Biochem. J. 2015, 469, 325–346. [Google Scholar] [CrossRef]
- Raffone, A.; Travaglino, A.; Cerbone, M.; De Luca, C.; Russo, D.; Di Maio, A.; De Marco, M.; Turco, M.C.; Insabato, L.; Zullo, F. Diagnostic accuracy of p53 immunohistochemistry as surrogate of TP53 sequencing in endometrial cancer. Pathol. Res. Pract. 2020, 216, 153025. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Luis, B.S.; Lane, D.P. Intratumour heterogeneity of p53 expression; causes and consequences. J. Pathol. 2019, 249, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Saft, L.; Karimi, M.; Ghaderi, M.; Matolcsy, A.; Mufti, G.J.; Kulasekararaj, A.; Göhring, G.; Giagounidis, A.; Selleslag, D.; Muus, P.; et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica 2014, 99, 1041–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Fernandez-Pol, S.; Ma, L.; Ohgami, R.S.; Arber, D.A. Immunohistochemistry for p53 is a useful tool to identify cases of acute myeloid leukemia with myelodysplasia-related changes that are TP53 mutated, have complex karyotype, and have poor prognosis. Mod. Pathol. 2016, 30, 382–392. [Google Scholar] [CrossRef]
- Tanner, S.D.; Baranov, V.I.; Ornatsky, O.I.; Bandura, D.R.; George, T.C. An introduction to mass cytometry: Fundamentals and applications. Cancer Immunol. Immunother. 2013, 62, 955–965. [Google Scholar] [CrossRef]
- Ornatsky, O.; Bandura, D.; Baranov, V.; Nitz, M.; Winnik, M.A.; Tanner, S. Highly multiparametric analysis by mass cytometry. J. Immunol. Methods 2010, 361, 1–20. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Nolan, G.P. Mass Cytometry: Single Cells, Many Features. Cell 2016, 165, 780–791. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, R.L.; Mak, D.H.; Burks, J.K.; Barton, M.C. Rapid monoisotopic cisplatin based barcoding for multiplexed mass cytometry. Sci. Rep. 2017, 7, 3779. [Google Scholar] [CrossRef] [Green Version]
- Zunder, E.R.; Lujan, E.; Goltsev, Y.; Wernig, M.; Nolan, G.P. A Continuous Molecular Roadmap to iPSC Reprogramming through Progression Analysis of Single-Cell Mass Cytometry. Cell Stem Cell 2015, 16, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, F.J.; Simonds, E.F.; Vivanco, N.; Bruce, T.; Borges, L.; Nolan, G.P.; Spitzer, M.H.; Bendall, S.C. Scalable Conjugation and Characterization of Immunoglobulins with Stable Mass Isotope Reporters for Single-Cell Mass Cytometry Analysis. Methods Mol. Biol. 2019, 1989, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Gullaksen, S.; Bader, L.; Hellesøy, M.; Sulen, A.; Fagerholt, O.H.E.; Engen, C.B.; Skavland, J.; Gjertsen, B.T.; Gavasso, S. Titrating Complex Mass Cytometry Panels. Cytom. Part A 2019, 95, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Fei, P.; El-Deiry, W.S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef] [Green Version]
- Haaland, I.; Opsahl, J.A.; Berven, F.S.; Reikvam, H.; Fredly, H.; Haugse, R.; Thiede, B.; McCormack, E.; Lain, S.; Bruserud, Ø.; et al. Molecular mechanisms of nutlin-3 involve acetylation of p53, histones and heat shock proteins in acute myeloid leukemia. Mol. Cancer 2014, 13, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allende-Vega, N.; Krzywinska, E.; Orecchioni, S.; López-Royuela, N.; Reggiani, F.; Talarico, G.; Rossi, J.-F.; Rossignol, R.; Hicheri, Y.; Cartron, G.; et al. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget 2015, 6, 19228–19245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, D.L.; Kabarowski, K.A.; Porubsky, V.L.; Kreeger, P.K. High-grade serous ovarian cancer cell lines exhibit heterogeneous responses to growth factor stimulation. Cancer Cell Int. 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Haines, D.S. Identification of a tumor-derived p53 mutant with novel transactivating selectivity. Oncogene 2000, 19, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.J.; Allan, G.J.; Shanahan, F.; Vousden, K.H.; Crook, T. p53 is frequently mutated in Burkitt’s lymphoma cell lines. Embo. J. 1991, 10, 2879–2887. [Google Scholar] [CrossRef]
- Cavalcanti, G.B.; Scheiner, M.A.M.; Magluta, E.P.S.; Vasconcelos, F.C.; Klumb, E.M.; Maia, R.C. p53 flow cytometry evaluation in leukemias: Correlation to factors affecting clinical outcome. Cytom. Part B Clin. Cytom. 2010, 78, 253–259. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, L.; Sun, L.-Q. The regulation of radiosensitivity by p53 and its acetylation. Cancer Lett. 2015, 363, 108–118. [Google Scholar] [CrossRef]
- Takahashi, C.; Au-Yeung, A.; Fuh, F.; Ramirez-Montagut, T.; Bolen, C.; Mathews, W.; O’Gorman, W.E. Mass cytometry panel optimization through the designed distribution of signal interference. Cytom. Part A 2016, 91, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Samusik, N.; Good, Z.; Spitzer, Z.G.M.H.; Davis, K.L.; Nolan, G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods 2016, 13, 493–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, E.-A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’Er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, A.D.; Gherardini, P.F.; Sykorova, M.; Yasin, F.; Nolan, G.P.; Slupsky, J.R.; Kalakonda, N. Multiplexed profiling of RNA and protein expression signatures in individual cells using flow or mass cytometry. Nat. Protoc. 2019, 14, 901–920. [Google Scholar] [CrossRef] [PubMed]
- Mavropoulos, A.; Allo, B.; He, M.; Park, E.; Majonis, D.; Ornatsky, O. Simultaneous Detection of Protein and mRNA in Jurkat and KG-1a Cells by Mass Cytometry. Cytom. Part A 2017, 91, 1200–1208. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.-C.; Surget, S.; Khoury, M.P. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. OncoTargets Ther. 2013, 7, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Okorokov, A.L.; Ponchel, F.; Milner, J. Induced N- and C-terminal cleavage of p53: A core fragment of p53, generated by interaction with damaged DNA, promotes cleavage of the N-terminus of full-length p53, whereas ssDNA induces C-terminal cleavage of p53. EMBO J. 1997, 16, 6008–6017. [Google Scholar] [CrossRef]
- Bonsing, B.A.; Corver, W.E.; Gorsira, M.C.; van Vliet, M.; Oud, P.S.; Cornelisse, C.J.; Fleuren, G.J. Specificity of seven monoclonal antibodies against p53 evaluated with Western blotting, immunohistochemistry, confocal laser scanning microscopy, and flow cytometry. Cytometry 1997, 28, 11–24. [Google Scholar] [CrossRef]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef]
- Allende-Vega, N.; Villalba, M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci. Rep. 2019, 9, 5637. [Google Scholar] [CrossRef]
- Leipold, M.D.; Obermoser, G.; Fenwick, C.; Kleinstuber, K.; Rashidi, N.; McNevin, J.P.; Nau, A.N.; Wagar, L.E.; Rozot, V.; Davis, M.M.; et al. Comparison of CyTOF assays across sites: Results of a six-center pilot study. J. Immunol. Methods 2018, 453, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Gaudilliere, B.; Angst, M.S.; Saeys, Y.; Aghaeepour, N. CytoNorm: A Normalization Algorithm for Cytometry Data. Cytom. Part A 2020, 97, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Bringeland, G.H.; Bader, L.; Blaser, N.; Budzinski, L.; Schulz, A.R.; Mei, H.E.; Myhr, K.-M.; Vedeler, C.A.; Gavasso, S. Optimization of Receptor Occupancy Assays in Mass Cytometry: Standardization Across Channels with QSC Beads. Cytom. Part A 2019, 95, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Nolan, G.P.; Roederer, M.; Chattopadhyay, P.K. A deep profiler’s guide to cytometry. Trends Immunol. 2012, 33, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maecker, H.T.; Harari, A. Immune monitoring technology primer: Flow and mass cytometry. J. Immunother. Cancer 2015, 3, 44. [Google Scholar] [CrossRef] [Green Version]
- Tricot, S.; Meyrand, M.; Sammicheli, C.; Elhmouzi-Younes, J.; Corneau, A.; Bertholet, S.; Malissen, M.; Le Grand, R.; Nuti, S.; Luche, H.; et al. Evaluating the efficiency of isotope transmission for improved panel design and a comparison of the detection sensitivities of mass cytometer instruments. Cytom. Part A 2015, 87, 357–368. [Google Scholar] [CrossRef]
- Irish, J.M.; Anensen, N.; Hovland, R.; Skavland, J.; Børresen-Dale, A.-L.; Bruserud, Ø.; Nolan, G.P.; Gjertsen, B.T.; Garand, R.; Lode, L.; et al. Flt3 Y591 duplication and Bcl-2 overexpression are detected in acute myeloid leukemia cells with high levels of phosphorylated wild-type p53. Blood 2006, 109, 2589–2596. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Famà, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef] [Green Version]
- Prochazka, K.T.; Pregartner, G.; Rücker, F.G.; Heitzer, E.; Pabst, G.; Wölfler, A.; Zebisch, A.; Berghold, A.; Döhner, K.; Sill, H. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica 2019, 104, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: Projections on diagnostic workup and therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Tuval, A.; Kaushansky, N.; Azogy, H.; Leshkowitz, D.; Salame, T.M.; Minden, M.D.; Tal, P.; Rotter, V.; Oren, M.; Shlush, L. Functional characterization of pre-leukemic hematopioetic cells. In Proceedings of the 25th EHA Congress—The European Hematology Association, Virtual Platform, The Hague, The Netherlands, 11–21 June 2020. [Google Scholar]
- Poláková, I.; Pelák, O.; Thürner, D.; Pokrývková, B.; Tachezy, R.; Kalina, T.; Smahel, M. Implementation of Mass Cytometry for Immunoprofiling of Patients with Solid Tumors. J. Immunol. Res. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leelatian, N.; Doxie, D.B.; Greenplate, A.R.; Mobley, B.C.; Lehman, J.M.; Sinnaeve, J.; Kauffmann, R.M.; Werkhaven, J.A.; Mistry, A.M.; Weaver, K.D.; et al. Single cell analysis of human tissues and solid tumors with mass cytometry. Cytom. Part B Clin. Cytom. 2016, 92, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Giesen, C.; Wang, H.A.O.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Rahman, A.H.; Tordesillas, L.; Berin, M.C. Heparin reduces nonspecific eosinophil staining artifacts in mass cytometry experiments. Cytom. Part A 2016, 89, 601–607. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell Type | p53 Status |
|---|---|---|
| MOLM13 | AML, M5 | Wild type |
| NB4 | AML, M3 | Missense mutation, R248Q [25] |
| OV-90 | Epithelial | Missense mutation, S215R [26] |
| Raji | Lymphoblast, Burkitt Lymphoma | Mutation, R213Q [27] and an Arg to Pro polymorphism at amino acid 72 [28] |
| HL-60 | AML, M2 | -/- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fagerholt, O.H.E.; Hellesøy, M.; Gullaksen, S.-E.; Gjertsen, B.T. Single Cell Detection of the p53 Protein by Mass Cytometry. Cancers 2020, 12, 3699. https://doi.org/10.3390/cancers12123699
Fagerholt OHE, Hellesøy M, Gullaksen S-E, Gjertsen BT. Single Cell Detection of the p53 Protein by Mass Cytometry. Cancers. 2020; 12(12):3699. https://doi.org/10.3390/cancers12123699
Chicago/Turabian StyleFagerholt, Oda Helen Eck, Monica Hellesøy, Stein-Erik Gullaksen, and Bjørn Tore Gjertsen. 2020. "Single Cell Detection of the p53 Protein by Mass Cytometry" Cancers 12, no. 12: 3699. https://doi.org/10.3390/cancers12123699
APA StyleFagerholt, O. H. E., Hellesøy, M., Gullaksen, S.-E., & Gjertsen, B. T. (2020). Single Cell Detection of the p53 Protein by Mass Cytometry. Cancers, 12(12), 3699. https://doi.org/10.3390/cancers12123699