DNA-Damage-Induced Alternative Splicing of p53

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Identification of Ribosomal Protein L26 (RPL26) as a Critical Mediator of p53 Translation after DNA Damage
2. Discovery of a DNA-Damage-Induced Alternative Splicing Pathway that Links the Beta Isoform of p53 (p53β) to Cellular Senescence
3. Post-Transcriptional Regulation of p53β
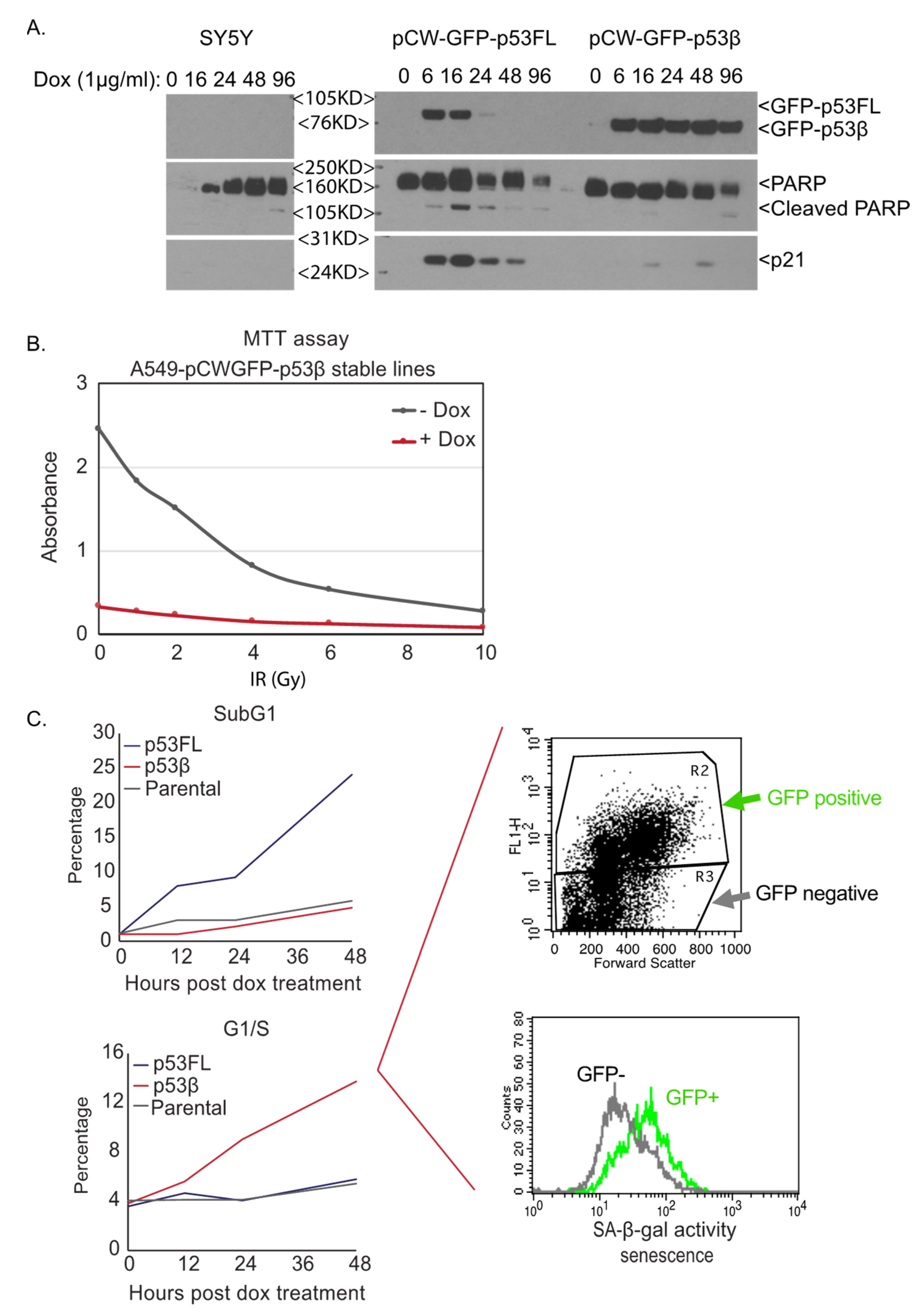
4. DNA Damage Induction of p53β Links to Cellular Senescence but not Apoptosis
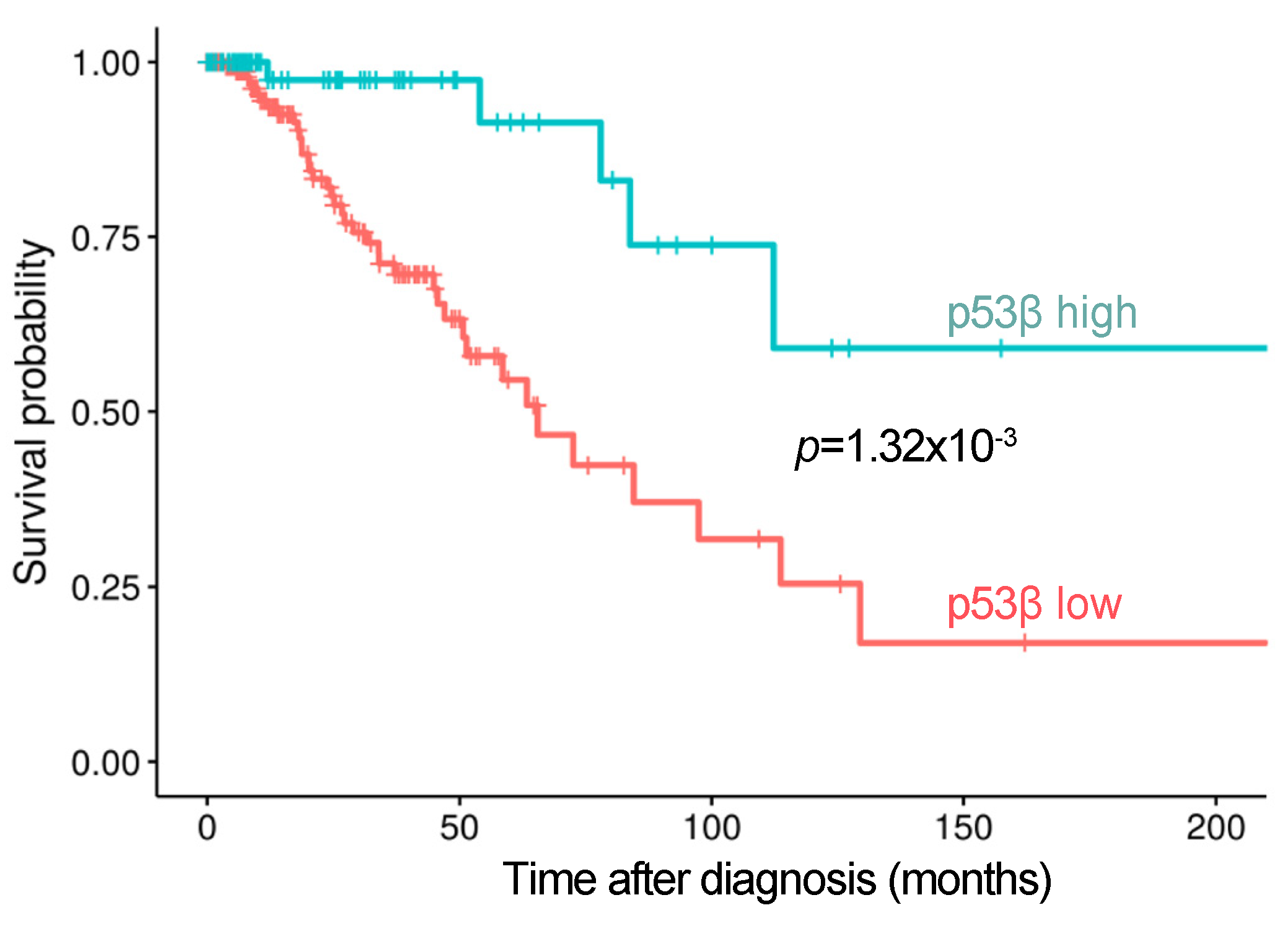
5. p53β Expression is Associated with Better Clinical Outcomes
6. Differential p53β Target Genes
7. Differential p53β Protein Interactors
8. Conclusions: Future Directions and Unanswered Questions
9. Patents
10. Materials and Methods:
10.1. Cell Culture, Stable Cell Lines, and Transfection
10.2. Plasmids and Chemicals
10.3. Immunoblot and Immunoprecipitation Using GFP-TRAP® and BioID
10.4. Flow SA-β-Galactosidase Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Kastan, M.B. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes. Dev. 2010, 24, 2146–2156. [Google Scholar] [CrossRef]
- Ofir-Rosenfeld, Y.; Boggs, K.; Michael, D.; Kastan, M.B.; Oren, M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 2008, 32, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Ashcroft, M.; Vousden, K.H. Regulation of p53 stability. Oncogene 1999, 18, 7637–7643. [Google Scholar] [CrossRef]
- Chen, J.; Guo, K.; Kastan, M.B. Interactions of nucleolin and ribosomal protein L26 (RPL26) in translational control of human p53 mRNA. J. Biol. Chem. 2012, 287, 16467–16476. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Chen, J.; Crutchley, J.; Zhang, D.; Owzar, K.; Kastan, M.B. Identification of a DNA Damage-Induced Alternative Splicing Pathway That Regulates p53 and Cellular Senescence Markers. Cancer Discov. 2017, 7, 766–781. [Google Scholar] [CrossRef]
- Canman, C.E.; Kastan, M.B. Small contribution of G1 checkpoint control manipulation to modulation of p53-mediated apoptosis. Oncogene 1998, 16, 957–966. [Google Scholar] [CrossRef][Green Version]
- Min, J.; Guo, K.; Suryadevara, P.K.; Zhu, F.; Holbrook, G.; Chen, Y.; Feau, C.; Young, B.M.; Lemoff, A.; Connelly, M.C.; et al. Optimization of a Novel Series of Ataxia-Telangiectasia Mutated Kinase Inhibitors as Potential Radiosensitizing Agents. J. Med. Chem. 2016, 59, 559–577. [Google Scholar] [CrossRef]
- Guo, K.; Shelat, A.A.; Guy, R.K.; Kastan, M.B. Development of a cell-based, high-throughput screening assay for ATM kinase inhibitors. J. Biomol. Screen. 2014, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes. Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.; Chabot, B. The emerging role of alternative splicing in senescence and aging. Aging Cell 2017, 16, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef]
- Marcel, V.; Fernandes, K.; Terrier, O.; Lane, D.P.; Bourdon, J.C. Modulation of p53beta and p53gamma expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 2014, 21, 1377–1387. [Google Scholar] [CrossRef]
- Cowen, L.E.; Luo, H.; Tang, Y. Characterization of SMG7 14-3-3-like domain reveals phosphoserine binding-independent regulation of p53 and UPF1. Sci. Rep. 2019, 9, 13097. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef]
- Yonish-Rouach, E.; Resnitzky, D.; Lotem, J.; Sachs, L.; Kimchi, A.; Oren, M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 1991, 352, 345–347. [Google Scholar] [CrossRef]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.M. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol. Biol. 1998, 79, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B. BioID: A screen for protein-protein interactions. Curr. Protoc Protein Sci. 2013, 74, 19.23.1–19.23.15. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Zhang, D.; Qin, X.; Owzar, K.; McCann, J.J.; Kastan, M.B. DNA-Damage-Induced Alternative Splicing of p53. Cancers 2021, 13, 251. https://doi.org/10.3390/cancers13020251
Chen J, Zhang D, Qin X, Owzar K, McCann JJ, Kastan MB. DNA-Damage-Induced Alternative Splicing of p53. Cancers. 2021; 13(2):251. https://doi.org/10.3390/cancers13020251
Chicago/Turabian StyleChen, Jing, Dadong Zhang, Xiaodi Qin, Kouros Owzar, Jennifer J. McCann, and Michael B. Kastan. 2021. "DNA-Damage-Induced Alternative Splicing of p53" Cancers 13, no. 2: 251. https://doi.org/10.3390/cancers13020251
APA StyleChen, J., Zhang, D., Qin, X., Owzar, K., McCann, J. J., & Kastan, M. B. (2021). DNA-Damage-Induced Alternative Splicing of p53. Cancers, 13(2), 251. https://doi.org/10.3390/cancers13020251