3′RNA Sequencing Accurately Classifies Formalin-Fixed Paraffin-Embedded Uterine Leiomyomas

 ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
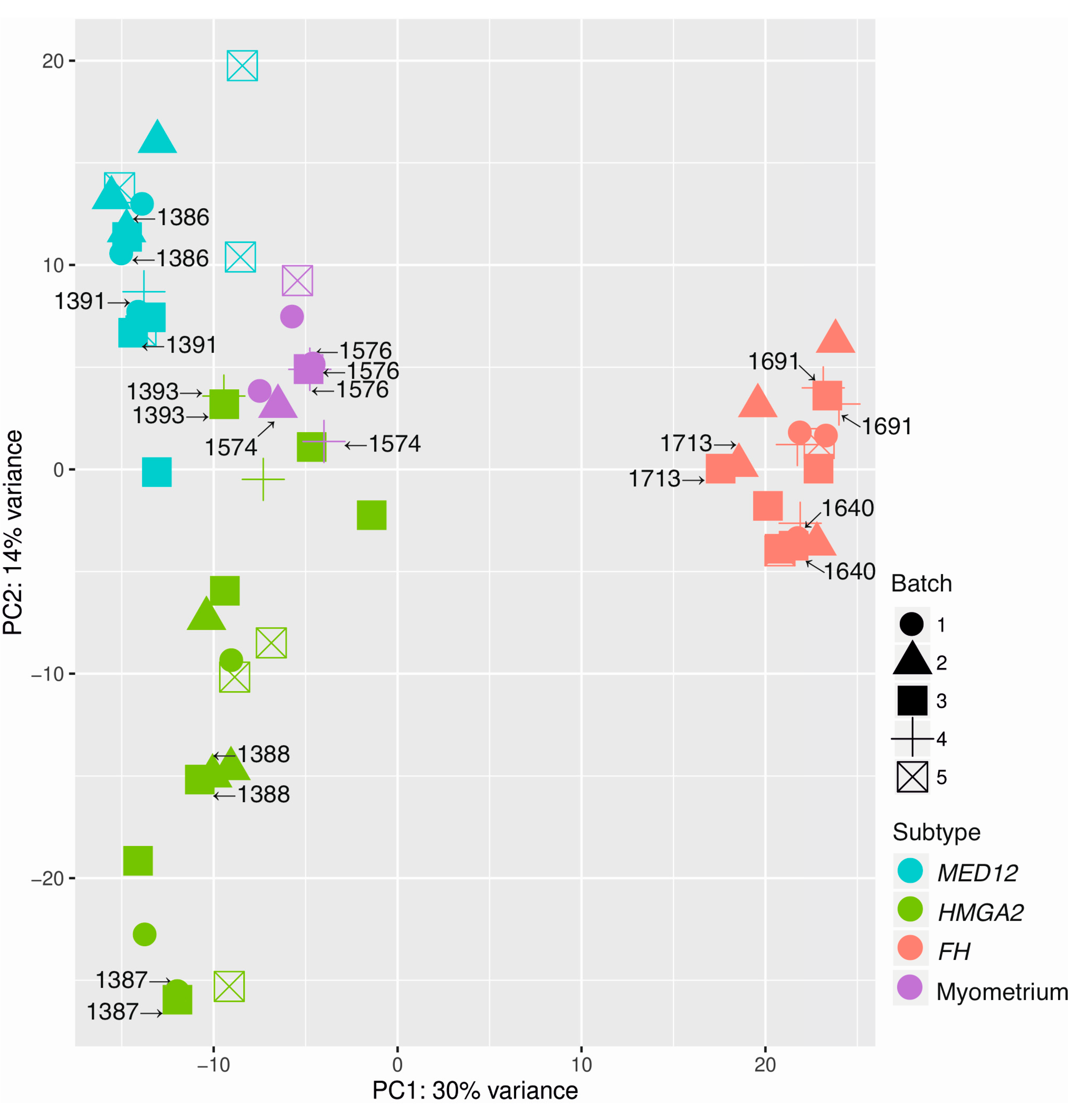
2.1. Principal Component Analysis Confirms Different Molecular Subtypes
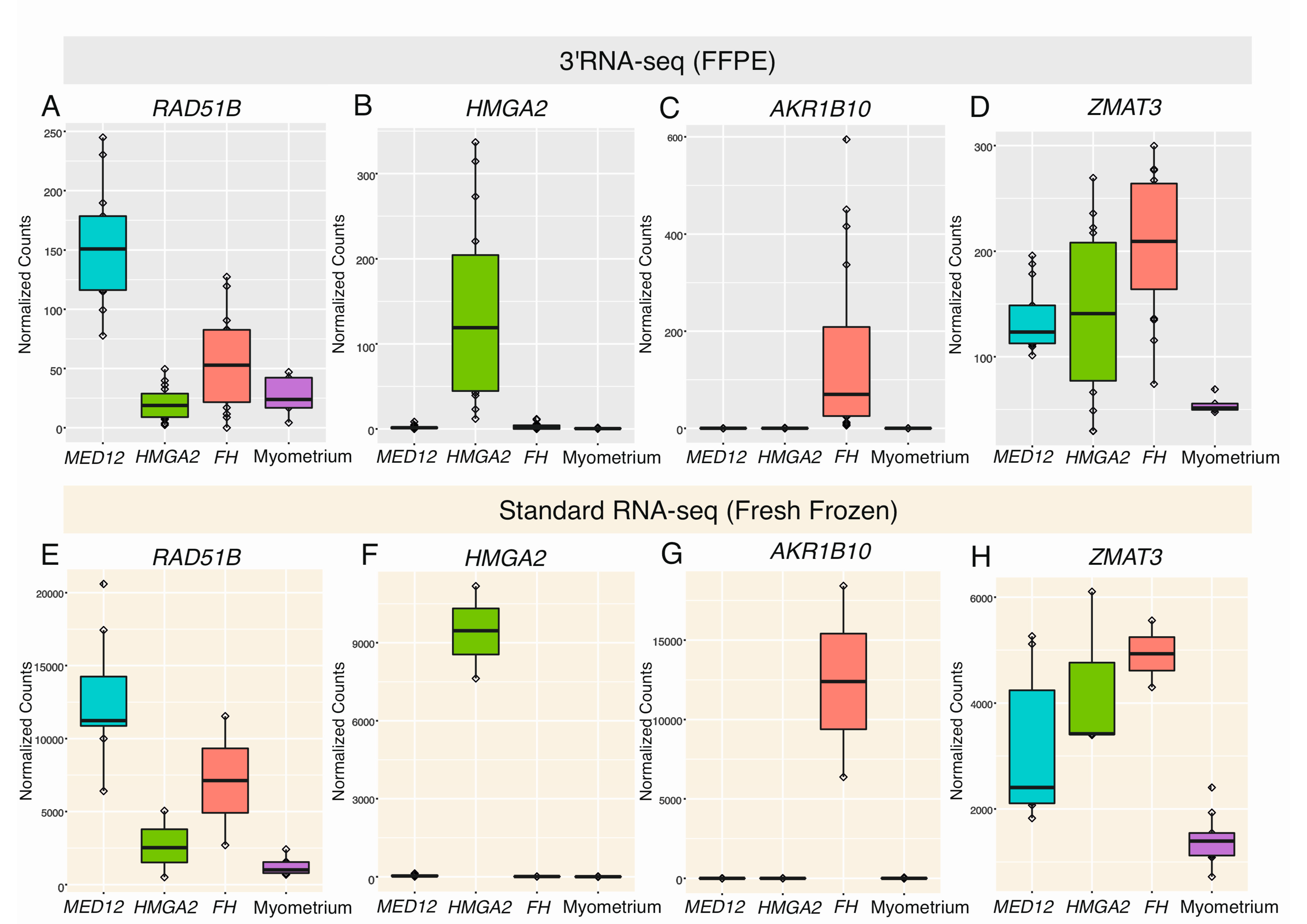
2.2. Differential Expression Analysis Confirms Distinct Gene Expression Patterns
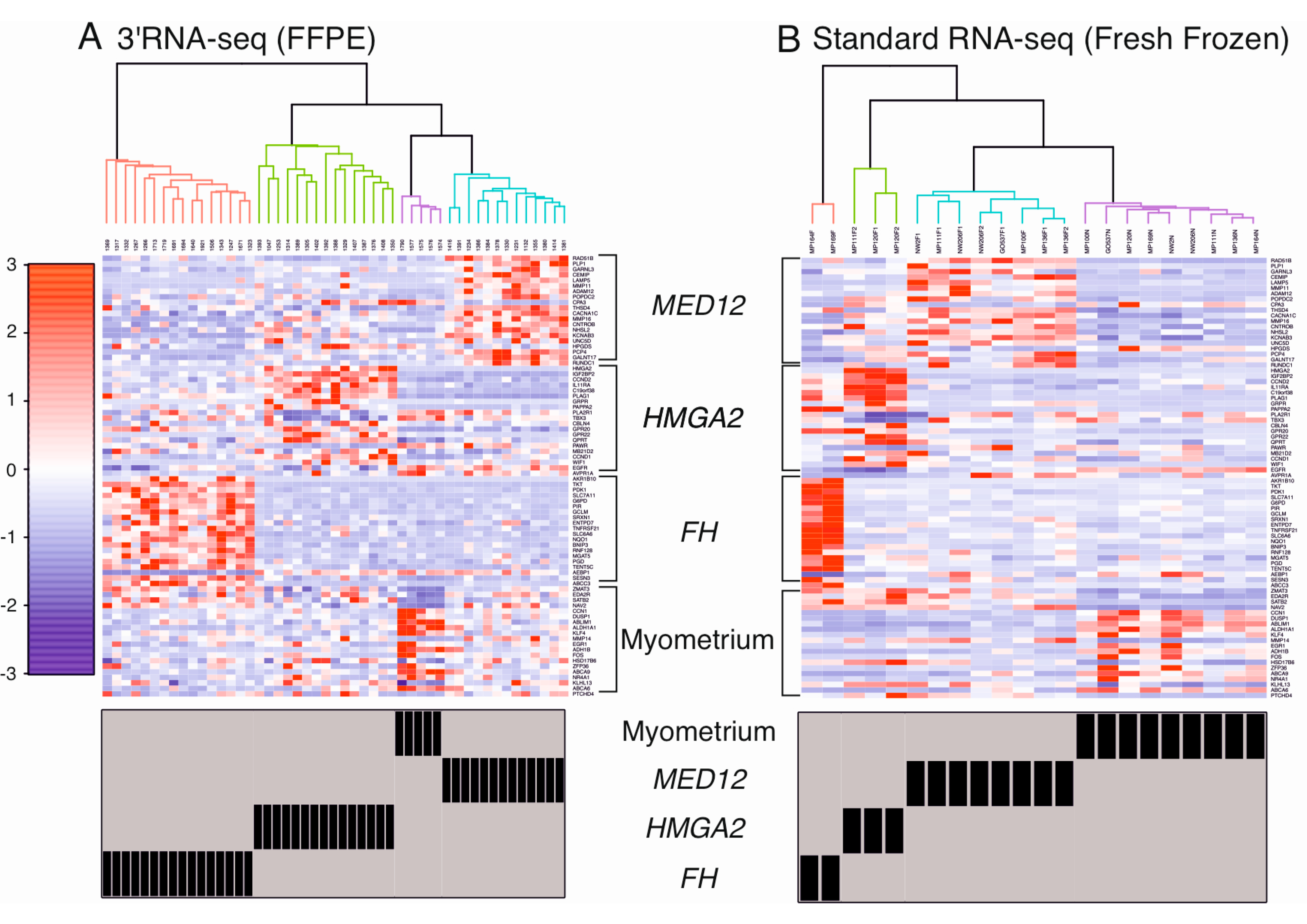
2.3. Supervised Hierarchical Clustering Accurately Classifies Leiomyomas
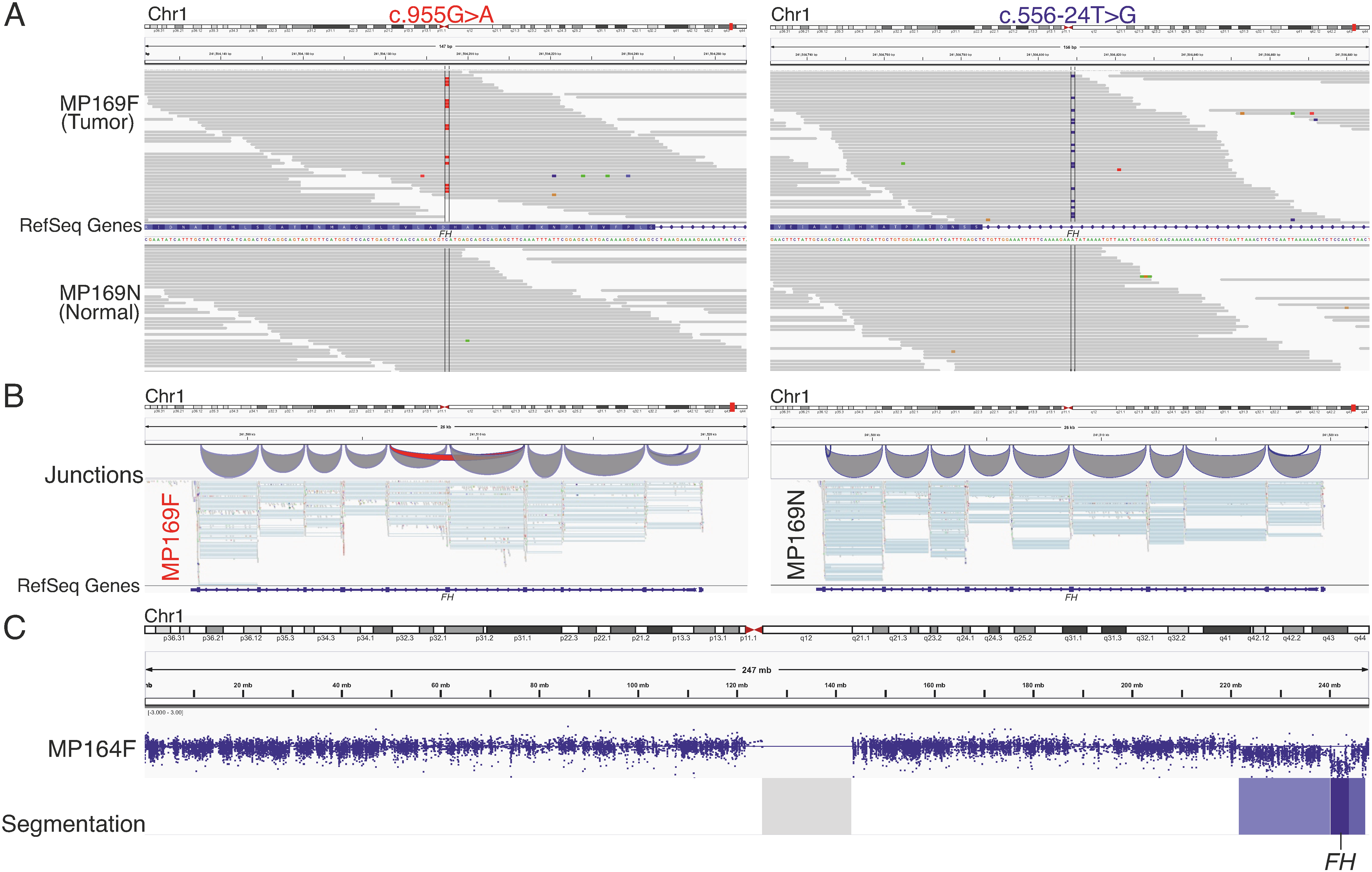
2.4. Exome Sequencing Reveals Biallelic Loss of FH in Two HMGA1 Overexpressing Leiomyomas
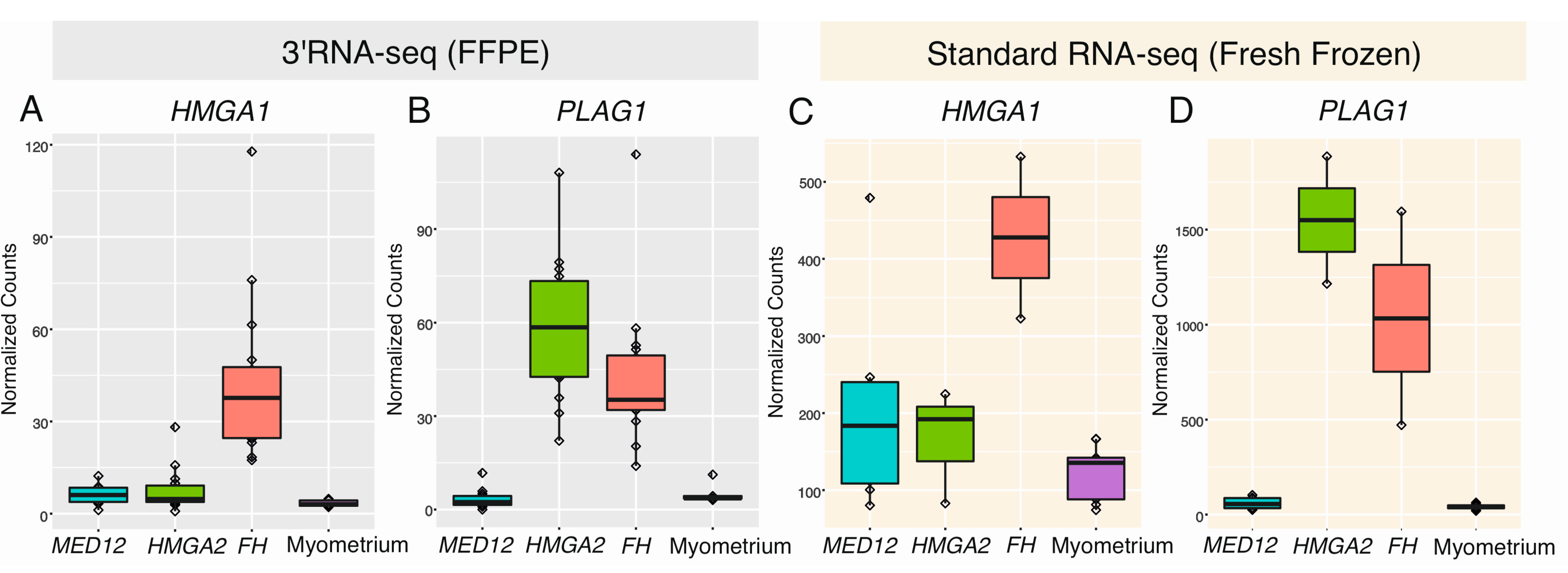
2.5. HMGA1 and PLAG1 Are Upregulated in Leiomyomas of the FH Subtype
3. Discussion
4. Materials and Methods
4.1. Study Material and Sample Selection
4.2. RNA Extraction and Sequencing
4.3. RNA Sequencing Data Analysis
4.4. Whole-Exome Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stewart, E.A.; Cookson, C.L.; Gandolfo, R.A.; Schulze-Rath, R. Epidemiology of uterine fibroids: A systematic review. BJOG Int. J. Obs. Gynaecol. 2017, 124, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Sabry, M.; Al-Hendy, A. Medical treatment of uterine leiomyoma. Reprod. Sci. 2012, 19, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.A. Uterine fibroids. N. Engl. J. Med. 2015, 372, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, S.; Young, R.H. WHO Classification of Tumours of Female Reproductive Organs, 4th ed.; IARC Publications: Lyon, France, 2014. [Google Scholar]
- Mehine, M.; Mäkinen, N.; Heinonen, H.; Aaltonen, L.A.; Vahteristo, P. Genomics of uterine leiomyomas: Insights from high-throughput sequencing. Fertil. Steril. 2014, 102, 621–629. [Google Scholar] [CrossRef]
- Lehtonen, H.J. Hereditary leiomyomatosis and renal cell cancer: Update on clinical and molecular characteristics. Fam. Cancer 2011, 10, 397–411. [Google Scholar] [CrossRef]
- Mehine, M.; Kaasinen, E.; Heinonen, H.; Mäkinen, N.; Kämpjärvi, K.; Sarvilinna, N.; Aavikko, M.; Vähärautio, A.; Pasanen, A.; Butzow, R.; et al. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc. Natl. Acad. Sci. USA 2016, 113, 1315–1320. [Google Scholar] [CrossRef]
- Panagopoulos, I.; Gorunova, L.; Brunetti, M.; Agostini, A.; Andersen, H.K.; Lobmaier, I.; Bjerkehagen, B.; Heim, S. Genetic heterogeneity in leiomyomas of deep soft tissue. Oncotarget 2017, 8, 48769–48781. [Google Scholar] [CrossRef][Green Version]
- Markowski, D.N.; Bartnitzke, S.; Loning, T.; Drieschner, N.; Helmke, B.M.; Bullerdiek, J. MED12 mutations in uterine fibroids–Their relationship to cytogenetic subgroups. Int. J. Cancer 2012, 131, 1528–1536. [Google Scholar] [CrossRef]
- Li, Y.; Qiang, W.; Griffin, B.B.; Gao, T.; Chakravarti, D.; Bulun, S.; Kim, J.J.; Wei, J. HMGA2-mediated tumorigenesis through angiogenesis in leiomyoma. Fertil. Steril. 2020, 114, 1085–1096. [Google Scholar] [CrossRef]
- Heinonen, H.; Pasanen, A.; Heikinheimo, O.; Tanskanen, T.; Palin, K.; Tolvanen, J.; Vahteristo, P.; Sjöberg, J.; Pitkänen, E.; Butzow, R.; et al. Multiple clinical characteristics separate MED12-mutation-positive and-negative uterine leiomyomas. Sci. Rep. 2017, 7, 1015. [Google Scholar] [CrossRef]
- Sandberg, A.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Leiomyoma. Cancer Genet. Cytogenet. 2005, 158, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, N.; Kämpjärvi, K.; Frizzell, N.; Butzow, R.; Vahteristo, P. Characterization of MED12, HMGA2, and FH alterations reveals molecular variability in uterine smooth muscle tumors. Mol. Cancer 2017, 16, 101. [Google Scholar] [CrossRef] [PubMed]
- George, J.W.; Fan, H.; Johnson, B.; Carpenter, T.J.; Foy, K.K.; Chatterjee, A.; Patterson, A.L.; Koeman, J.; Adams, M.; Madaj, Z.B.; et al. Integrated epigenome, exome, and transcriptome analyses reveal molecular subtypes and homeotic transformation in uterine fibroids. Cell Rep. 2019, 29, 4069–4085.e6. [Google Scholar] [CrossRef]
- Bardella, C.; El-Bahrawy, M.; Frizzell, N.; Adam, J.; Ternette, N.; Hatipoglu, E.; Howarth, K.; O’Flaherty, L.; Roberts, I.; Turner, G.; et al. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 2011, 225, 4–11. [Google Scholar] [CrossRef]
- Kokkat, T.J.; Patel, M.S.; McGarvey, D.; LiVolsi, V.A.; Baloch, Z.W. Archived formalin-fixed paraffin-embedded (FFPE) blocks: A valuable underexploited resource for extraction of DNA, RNA, and protein. Biopreserv. Biobank. 2013, 11, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA quality from FFPE samples. PLoS ONE 2007, 2, e1261. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.; Sorokin, M.; Garazha, A.; Glusker, A.; Aleshin, A.; Poddubskaya, E.; Sekacheva, M.; Kim, E.; Gaifullin, N.; Giese, A.; et al. RNA sequencing for research and diagnostics in clinical oncology. Semin. Cancer Biol. 2020, 60, 311–323. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Moll, P.; Ante, M.; Seitz, A.; Reda, T. QuantSeq 3’ mRNA sequencing for RNA quantification. Nat. Methods 2014, 11, i–iii. [Google Scholar] [CrossRef]
- Lohman, B.K.; Weber, J.N.; Bolnick, D.I. Evaluation of TagSeq, a reliable low-cost alternative for RNAseq. Mol. Ecol. Resour. 2016, 16, 1315–1321. [Google Scholar] [CrossRef]
- Turnbull, A.K.; Selli, C.; Martinez-Perez, C.; Fernando, A.; Renshaw, L.; Keys, J.; Figueroa, J.D.; He, X.; Tanioka, M.; Munro, A.F.; et al. Unlocking the transcriptomic potential of formalin-fixed paraffin embedded clinical tissues: Comparison of gene expression profiling approaches. BMC Bioinform. 2020, 21, 30. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Klemke, M.; Muller, M.H.; Wosniok, W.; Markowski, D.N.; Nimzyk, R.; Helmke, B.M.; Bullerdiek, J. Correlated expression of HMGA2 and PLAG1 in thyroid tumors, uterine leiomyomas and experimental models. PLoS ONE 2014, 9, e88126. [Google Scholar] [CrossRef]
- Mäkinen, N.; Vahteristo, P.; Kämpjärvi, K.; Arola, J.; Butzow, R.; Aaltonen, L.A. MED12 exon 2 mutations in histopathological uterine leiomyoma variants. Eur. J. Hum. Genet. 2013, 21, 1300–1303. [Google Scholar] [CrossRef]
- Kämpjärvi, K.; Mäkinen, N.; Mehine, M.; Välipakka, S.; Uimari, O.; Pitkänen, E.; Heinonen, H.; Heikkinen, T.; Tolvanen, J.; Ahtikoski, A.; et al. MED12 mutations and FH inactivation are mutually exclusive in uterine leiomyomas. Br. J. Cancer 2016, 114, 1405–1411. [Google Scholar] [CrossRef]
- Ahvenainen, T.V.; Mäkinen, N.M.; von Nandelstadh, P.; Vahteristo, M.E.A.; Pasanen, A.M.; Bützow, R.C.; Vahteristo, P.M. Loss of ATRX/DAXX expression and alternative lengthening of telomeres in uterine leiomyomas. Cancer 2018, 124, 4650–4656. [Google Scholar] [CrossRef]
- Äyräväinen, A.; Pasanen, A.; Ahvenainen, T.; Heikkinen, T.; Pakarinen, P.; Härkki, P.; Vahteristo, P. Systematic molecular and clinical analysis of uterine leiomyomas from fertile-aged women undergoing myomectomy. Hum. Reprod. 2020, 35, 2237–2244. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemelä, P.; Gentile, M.; Scheinin, I.; Koski, M.; Käki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genom. 2011, 12, 507. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Katainen, R.; Donner, I.; Cajuso, T.; Kaasinen, E.; Palin, K.; Mäkinen, V.; Aaltonen, L.A.; Pitkänen, E. Discovery of potential causative mutations in human coding and noncoding genome with the interactive software BasePlayer. Nat. Protoc. 2018, 13, 2580–2600. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehine, M.; Khamaiseh, S.; Ahvenainen, T.; Heikkinen, T.; Äyräväinen, A.; Pakarinen, P.; Härkki, P.; Pasanen, A.; Bützow, R.; Vahteristo, P. 3′RNA Sequencing Accurately Classifies Formalin-Fixed Paraffin-Embedded Uterine Leiomyomas. Cancers 2020, 12, 3839. https://doi.org/10.3390/cancers12123839
Mehine M, Khamaiseh S, Ahvenainen T, Heikkinen T, Äyräväinen A, Pakarinen P, Härkki P, Pasanen A, Bützow R, Vahteristo P. 3′RNA Sequencing Accurately Classifies Formalin-Fixed Paraffin-Embedded Uterine Leiomyomas. Cancers. 2020; 12(12):3839. https://doi.org/10.3390/cancers12123839
Chicago/Turabian StyleMehine, Miika, Sara Khamaiseh, Terhi Ahvenainen, Tuomas Heikkinen, Anna Äyräväinen, Päivi Pakarinen, Päivi Härkki, Annukka Pasanen, Ralf Bützow, and Pia Vahteristo. 2020. "3′RNA Sequencing Accurately Classifies Formalin-Fixed Paraffin-Embedded Uterine Leiomyomas" Cancers 12, no. 12: 3839. https://doi.org/10.3390/cancers12123839
APA StyleMehine, M., Khamaiseh, S., Ahvenainen, T., Heikkinen, T., Äyräväinen, A., Pakarinen, P., Härkki, P., Pasanen, A., Bützow, R., & Vahteristo, P. (2020). 3′RNA Sequencing Accurately Classifies Formalin-Fixed Paraffin-Embedded Uterine Leiomyomas. Cancers, 12(12), 3839. https://doi.org/10.3390/cancers12123839