A Machine-Learning Tool Concurrently Models Single Omics and Phenome Data for Functional Subtyping and Personalized Cancer Medicine



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
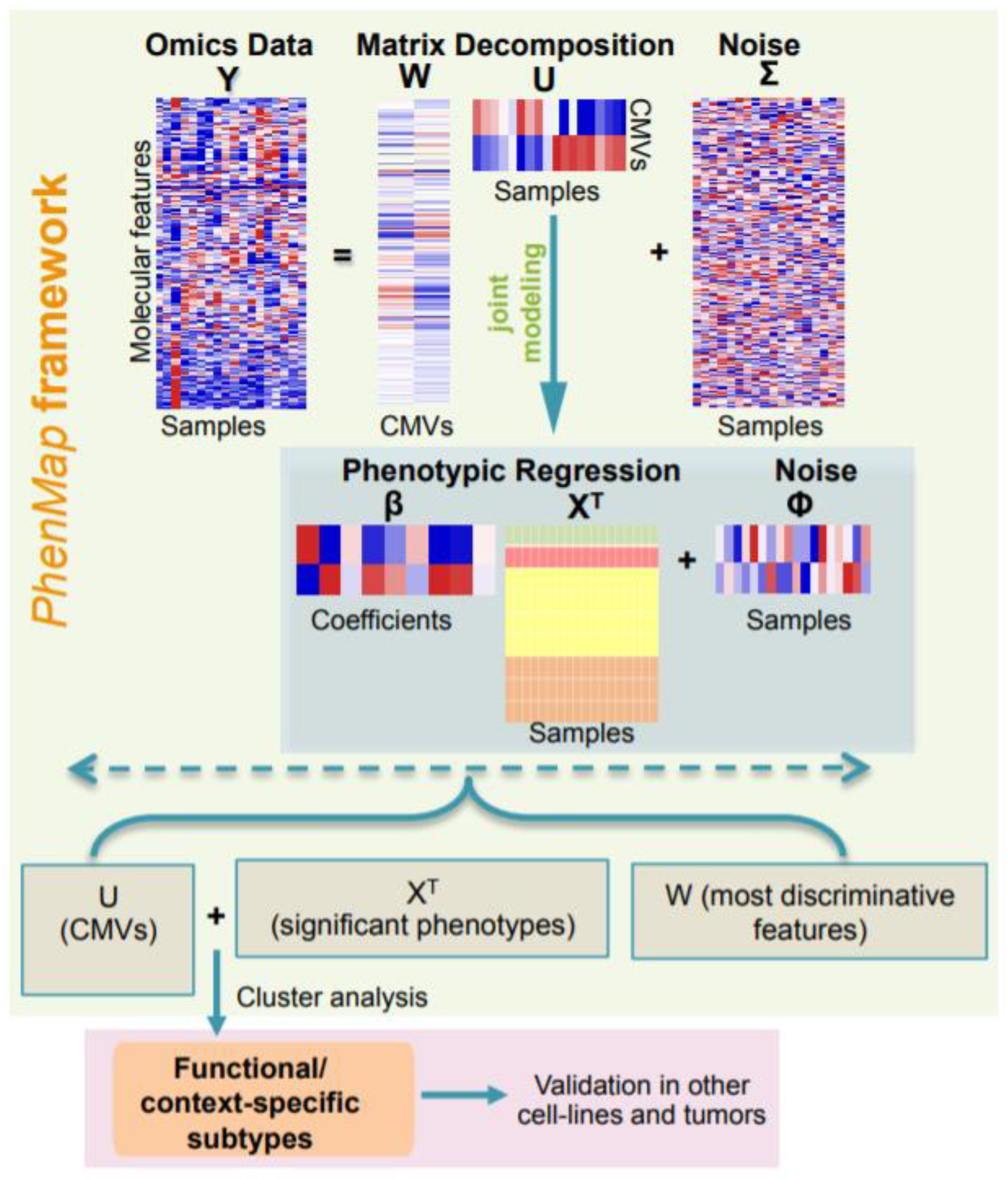
2.1. Overview of PhenMap
2.2. Example 1
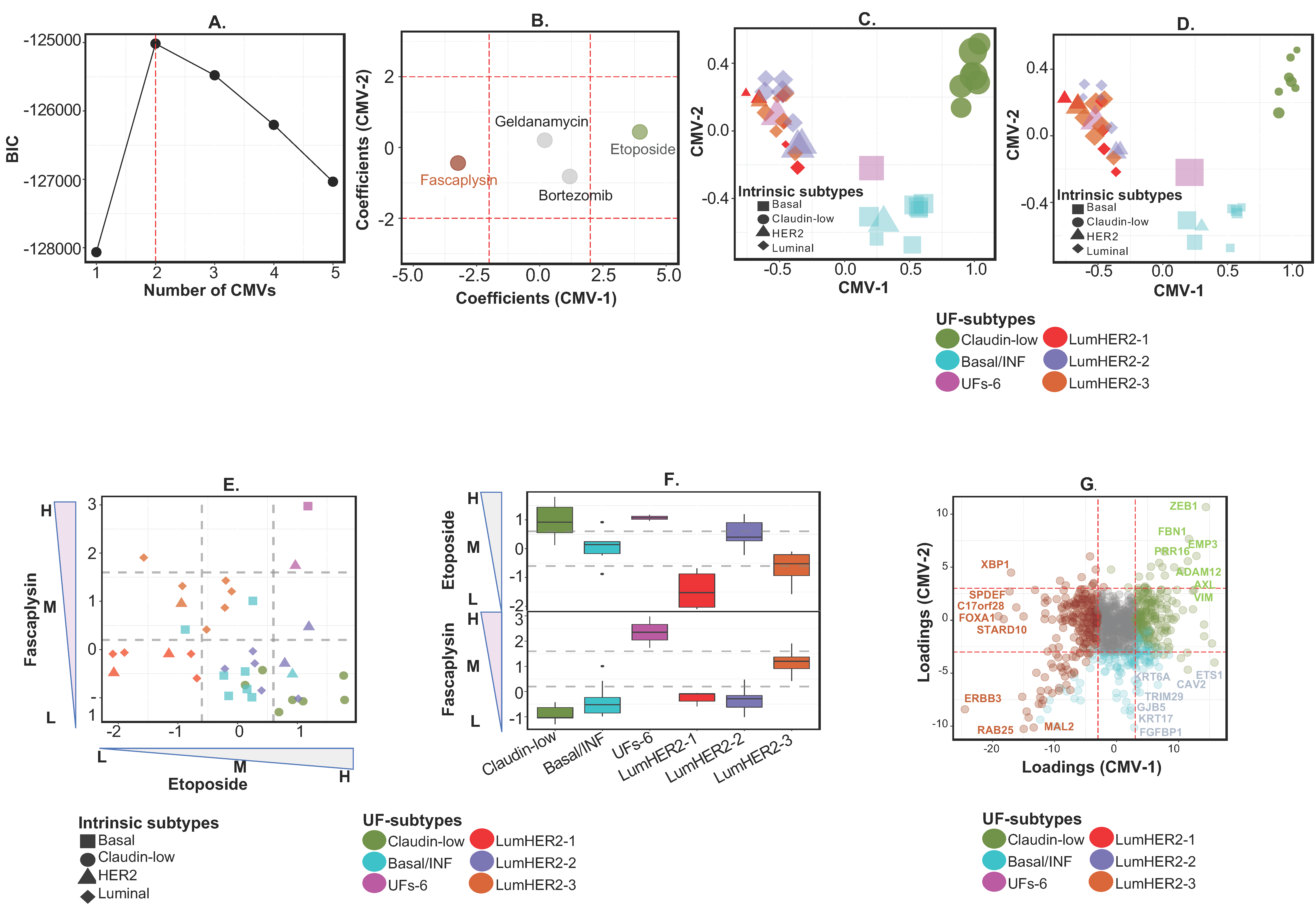
2.2.1. Example 1A—Identifying Functional and Discrete Subtypes in Breast Cancer with Drug Response Biomarkers
2.2.2. Example 1B—Identifying “Context-Specific” Functional Subtypes in Breast Cancer Cell Lines
2.3. Example 2—Identifying Continuous or Discrete Subtypes with Clinical Implications by Associating CMVs with Phenotypes Using PhenMap
3. Methods
3.1. PhenMap
3.2. Sparseness and Prior Distributions Associated with Features and Phenotypes
3.3. Model Selection
3.4. Model Convergence and Fitting in PhenMap
3.5. The Algorithm in PhenMap
- (1)
- Derive the CMVs, U(s), from a Gaussian full conditional distribution,
- (2)
- Derive the precision hyper-parameters Λ(s) from a Gamma full conditional distribution,
- (3)
- Derive the loadings matrix W(s) from a Gaussian full conditional distribution,
- (4)
- Derive the error covariance Σ(s) from an inverse-Gamma full conditional distribution,
- (5)
- Derive the regression coefficients β(s) from a Gaussian full conditional distribution, and
- (6)
- Derive the CMV covariance parameters Φ(s) from an inverse-Gamma full conditional distribution.
3.6. Clustering of Context-Specific Phenotypic Mapping Variables (CMVs) with Drug Response Information
3.7. Development of Classifiers
3.8. Datasets and Samples
3.9. Availability of Data and Material
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
References
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Gonzalez Ostos, L.C. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grötzinger, C.; Barbi, S.; Bersani, S.; Korner, J.; Wafy, I.; Mafficini, A.; Lawlor, R.T.; et al. A cross-species analysis in pancreatic neuroendocrine tumors reveals molecular subtypes with distinctive clinical, metastatic, developmental, and metabolic characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moore, A. K-means and Hierarchical Clustering. Stat. Data Min. Tutor. 2001, 47, 1–24. [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Fontana, E.; Eason, K.; Cervantes, A.R.S.; Sadanandam, A. Context matters—Consensus molecular subtypes of colorectal cancer as biomarkers for clinical trials. Ann. Oncol. 2019, 30, 520–527. [Google Scholar] [CrossRef]
- Pusztai, L.; Mazouni, C.; Anderson, K.; Wu, Y.; Symmans, W. Molecular classification of breast cancer: Limitations and potential. Oncologist 2006, 11, 868–877. [Google Scholar] [CrossRef]
- Janice, L. Palbociclib: A first-in-class CDK4/CDK6 inhibitorfor the treatment of hormone-receptor positiveadvanced breast cancer. J. Hematol. Oncol. 2015, 8, 98. [Google Scholar]
- Heiser, L.M.; Sadanandam, A.; Kuo, W.-L.; Benz, S.C.; Goldstein, T.C.; Ng, S.; Gibb, W.J.; Wang, N.J.; Ziyad, S.; Tong, F.; et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poudel, P.; Nyamundanda, G.; Patil, Y.; Cheang, M.C.U.; Sadanandam, A. Heterocellular gene signatures reveal luminal-A breast cancer heterogeneity and differential therapeutic responses. NPJ Breast Cancer 2019, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Sinha, R.; Hoadley, K.A.; Jacobsen, A.S.; Reva, B.; Perou, C.M.; Sander, C.; Schultz, N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res. Treat. 2013, 141, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Giorgetti, C. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.; Madden, S.F.; Doolan, P.; Aherne, S.T.; Joyce, H.; O’Driscoll, L.; Kennedy, S. Correlating transcriptional networks to breast cancer survival: A large-scale coexpression analysis. Carcinogenesis 2013, 34, 2300–2308. [Google Scholar] [CrossRef]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef] [Green Version]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [Green Version]
- Engelhardt, B.E.; Stephens, M. Analysis of population structure: A unifying framework and novel methods based on sparse factor analysis. PLoS Genet. 2010, 6, e1001117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.; Bottolo, L.; Al, E. Bayesian models for sparse regression analysis of high dimensional data. Bayesian Stat. 2010, 9, 539–569. [Google Scholar]
- Hoff, P. A First Course in Bayesian Statistical Methods; Springer: New York, NY, USA, 2009. [Google Scholar]
- Schwarz, G.E. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Fraley, C.; Raftery, A.E. Bayesian Regularization for Normal Mixture Estimation and Model-Based Clustering. J. Classif. 2007, 24, 155–181. [Google Scholar] [CrossRef] [Green Version]
- Costa, I.G.; Roepcke, S.; Hafemeister, C.; Schliep, A. Inferring differentiation pathways from gene expression. Bioinformatics 2008, 24, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Ishwaran, H.; Rao, J.S. Spike and slab variable selection: Frequentist and bayesian strategies. Ann. Stat. 2005, 33, 730–773. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A.; Carlin, J.B.; Stern, H.S.; Rubin, D.B. Bayesian Data Analysis; Chapman and Hall/CRC: Boca Raton, FL, USA, 2003. [Google Scholar]
- Ansari, A.; Jedidi, K.; Dube, L. Heterogeneous factor analysis model: A Bayesian approach. Psychometrika 2002, 67, 49–78. [Google Scholar] [CrossRef]
- Gilks, W.R.; Richardson, S.; Spiegelhalter, D.J. Markov Chain Monte Carlo in Practice; Chapman and Hall: London, UK, 1996. [Google Scholar]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [Green Version]
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyamundanda, G.; Eason, K.; Guinney, J.; Lord, C.J.; Sadanandam, A. A Machine-Learning Tool Concurrently Models Single Omics and Phenome Data for Functional Subtyping and Personalized Cancer Medicine. Cancers 2020, 12, 2811. https://doi.org/10.3390/cancers12102811
Nyamundanda G, Eason K, Guinney J, Lord CJ, Sadanandam A. A Machine-Learning Tool Concurrently Models Single Omics and Phenome Data for Functional Subtyping and Personalized Cancer Medicine. Cancers. 2020; 12(10):2811. https://doi.org/10.3390/cancers12102811
Chicago/Turabian StyleNyamundanda, Gift, Katherine Eason, Justin Guinney, Christopher J. Lord, and Anguraj Sadanandam. 2020. "A Machine-Learning Tool Concurrently Models Single Omics and Phenome Data for Functional Subtyping and Personalized Cancer Medicine" Cancers 12, no. 10: 2811. https://doi.org/10.3390/cancers12102811
APA StyleNyamundanda, G., Eason, K., Guinney, J., Lord, C. J., & Sadanandam, A. (2020). A Machine-Learning Tool Concurrently Models Single Omics and Phenome Data for Functional Subtyping and Personalized Cancer Medicine. Cancers, 12(10), 2811. https://doi.org/10.3390/cancers12102811