BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
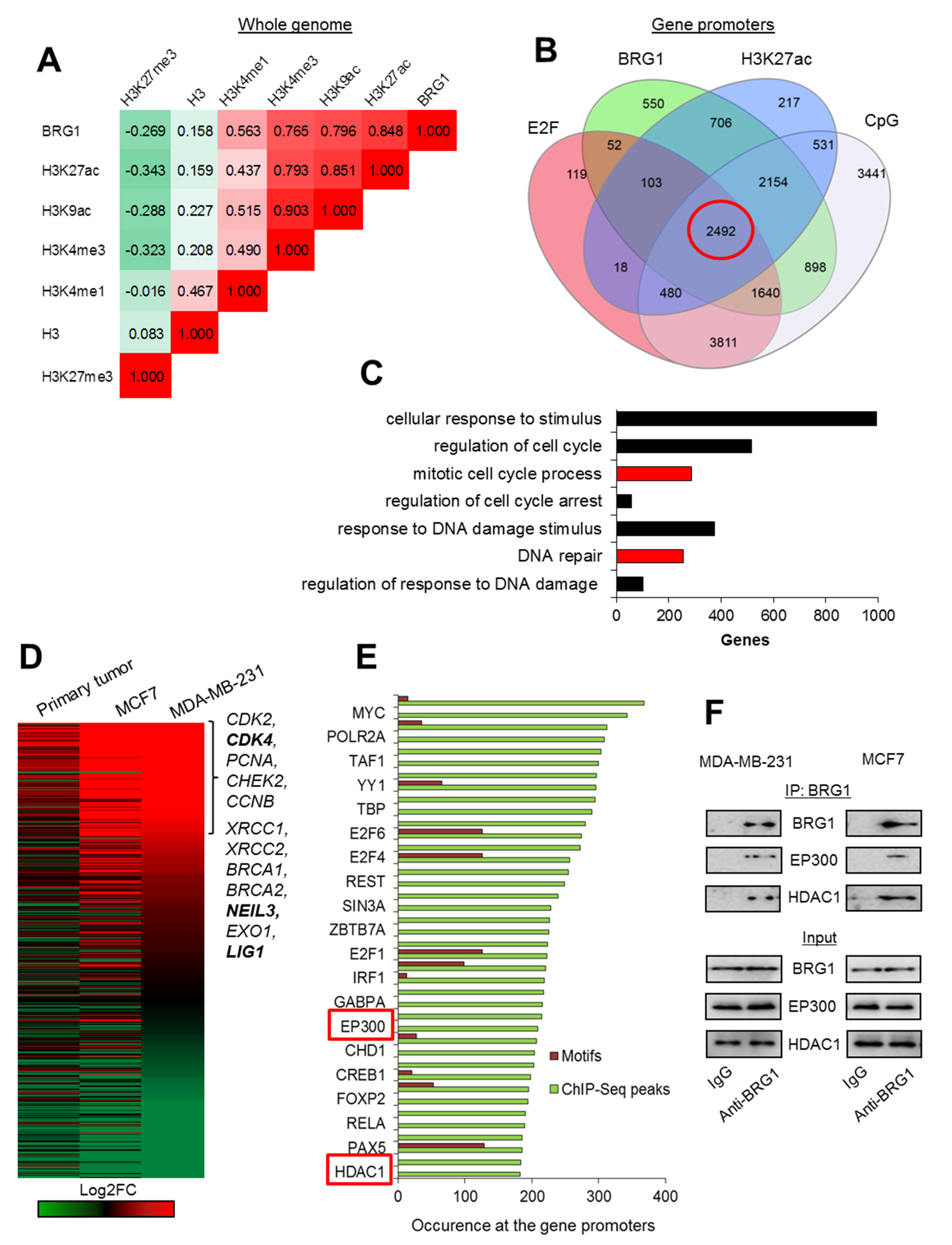
2.1. E2F/CpG Motifs at the Acetylated Gene Promoters Mark BRG1 Distribution in Genome of Breast Cancer Cells
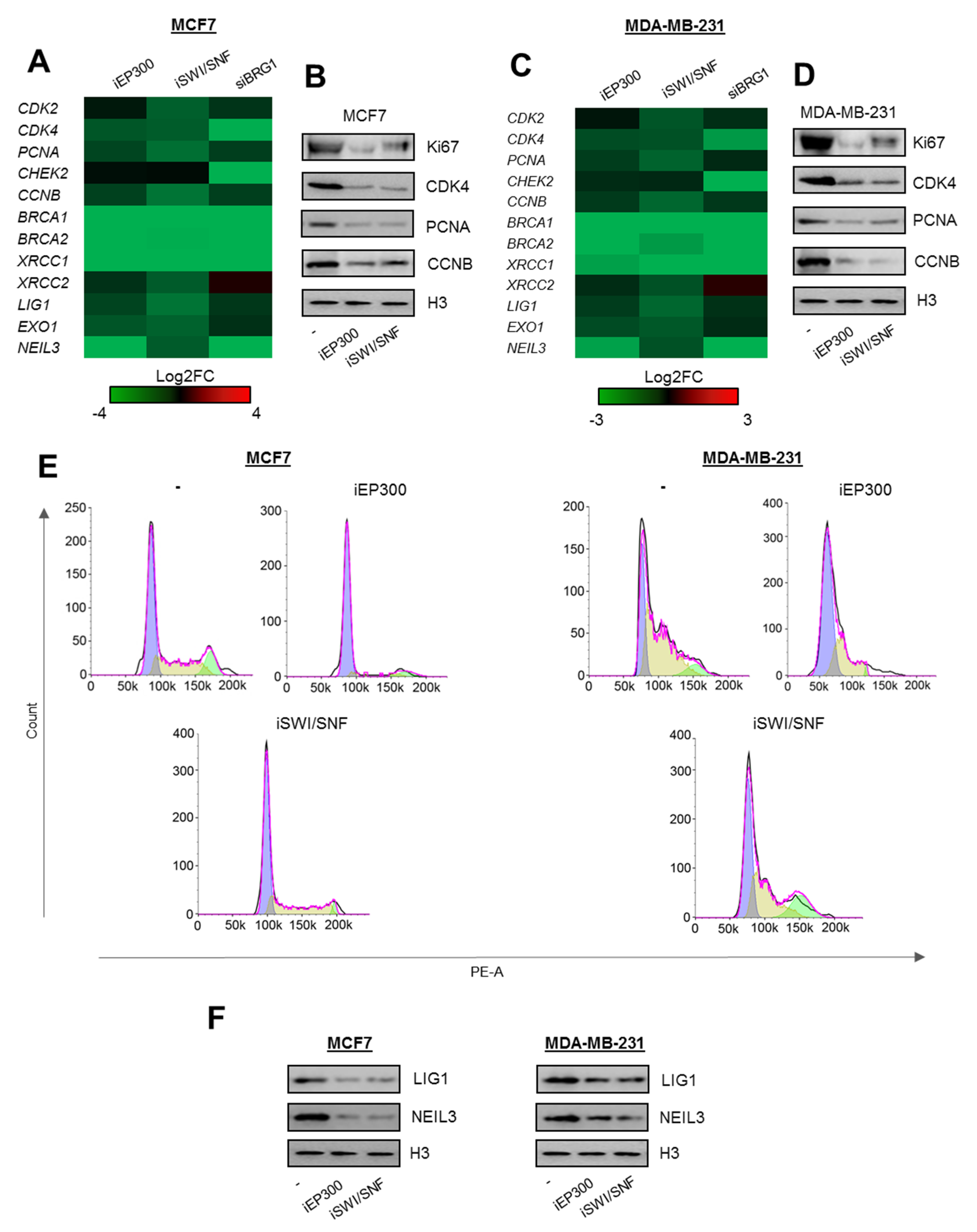
2.2. BRG1-EP300 Complexes Drive Transcription of Some Proliferation and DNA Repair Genes
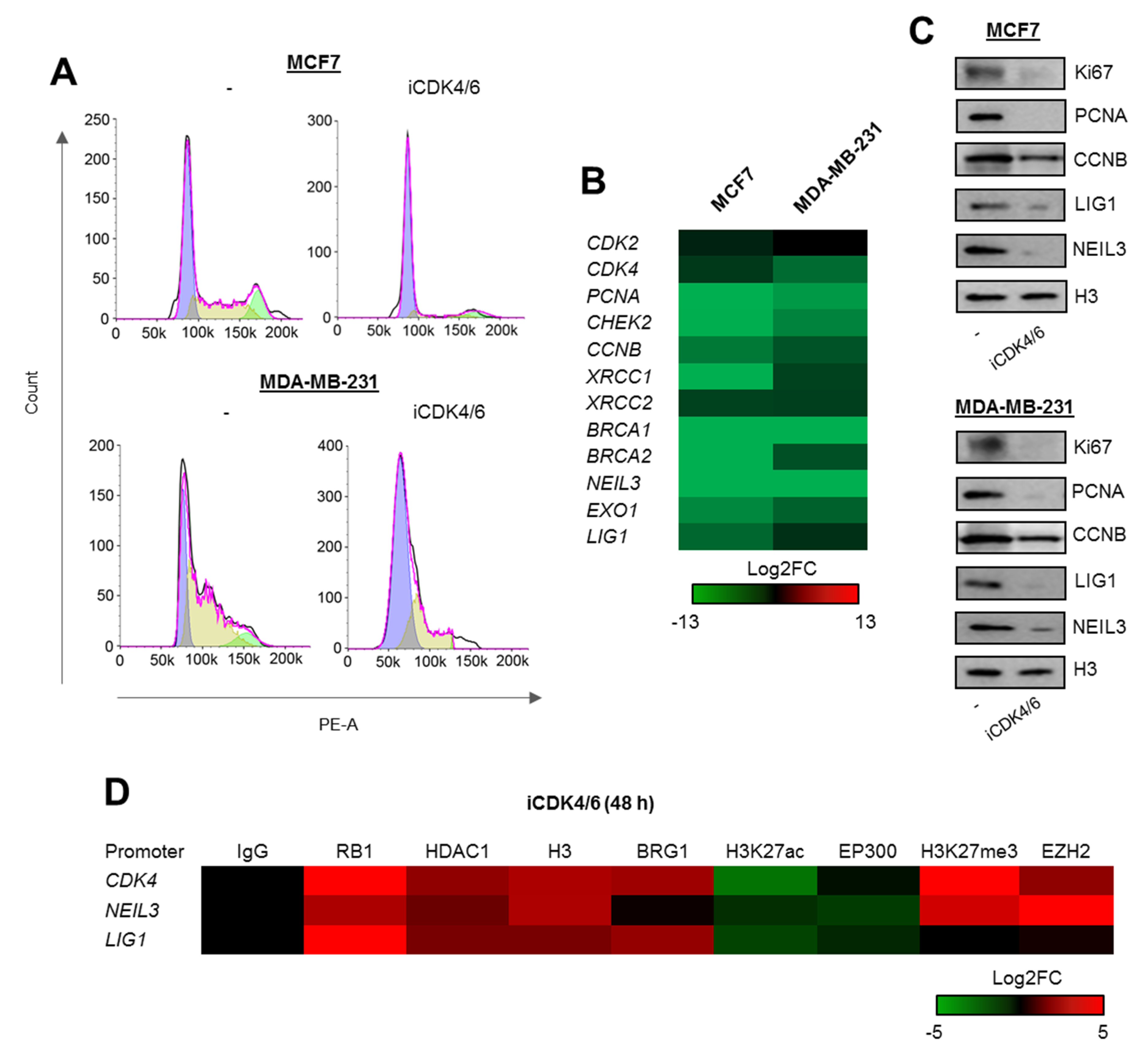
2.3. Cell Cycle-Dependent Chromatin Composition Controls Transcription of Proliferation and DNA Repair Genes
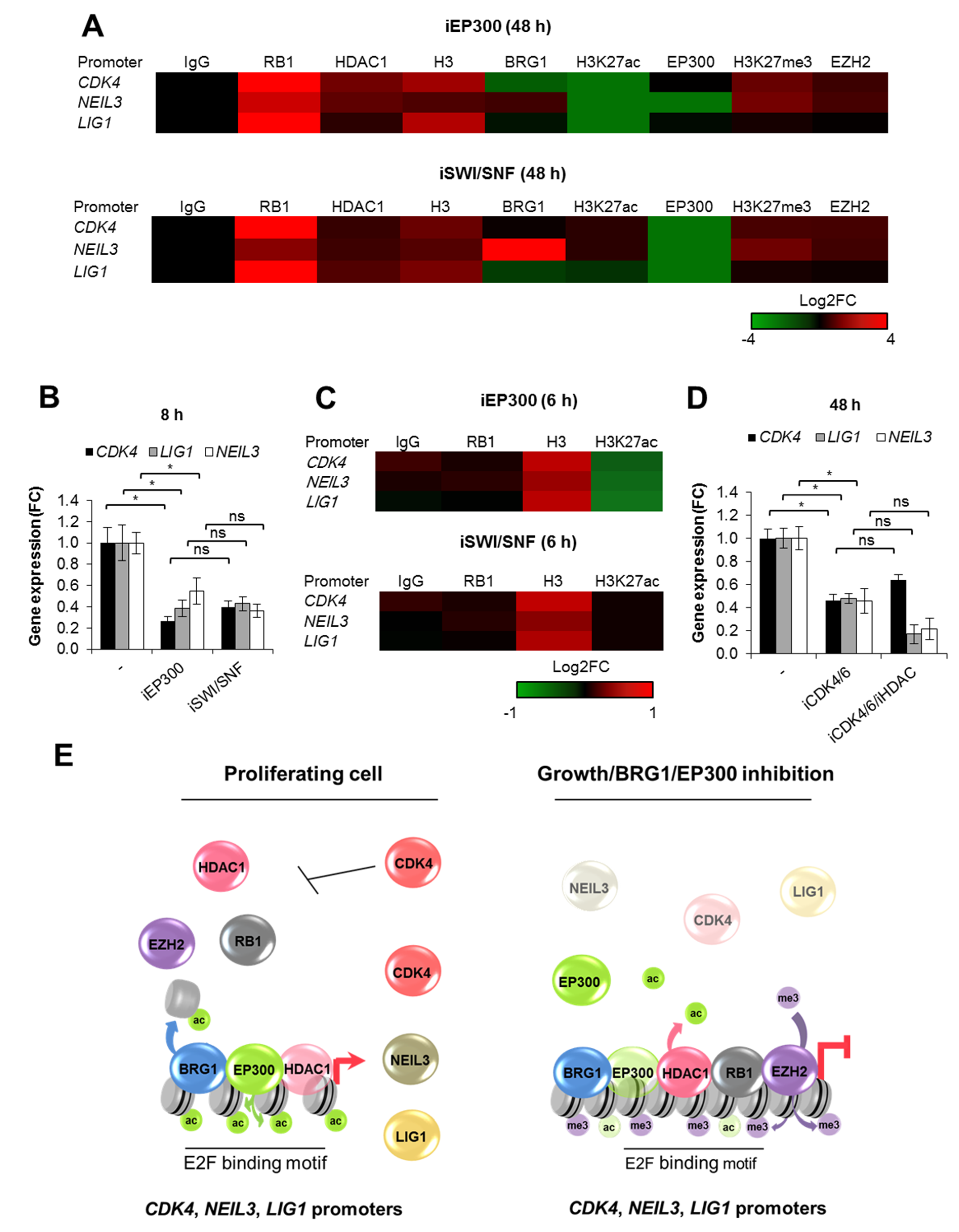
2.4. BRG1 Couples Cell Divisions with Transcription of DNA Repair Genes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Treatment with Inhibitors
4.3. Quantification of Gene Expression
4.4. Evaluation of Cell Proliferation
4.5. Chromatin Immunoprecipitation
4.6. Transient Gene Silencing
4.7. ChIP-Seq Analysis in Galaxy Version 19.05.dev
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Selfors, L.M.; Stover, D.G.; Harris, I.S.; Brugge, J.S.; Coloff, J.L. Identification of cancer genes that are independent of dominant proliferation and lineage programs. Proc. Natl. Acad. Sci. USA 2017, 114, E11276–E11284. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, J.; Ploszaj, T.; Pulaski, L.; Robaszkiewicz, A. EP300-HDAC1-SWI/SNF functional unit defines transcription of some DNA repair enzymes during differentiation of human macrophages. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Saladi, S.V.; Basuroy, T.; Marathe, H.G.; Lores, P.; de la Serna, I.L. BAF60A mediates interactions between the microphthalmia-associated transcription factor and the BRG1-containing SWI/SNF complex during melanocyte differentiation. J. Cell Physiol. 2019, 234, 11780–11791. [Google Scholar] [CrossRef] [PubMed]
- Raab, J.R.; Runge, J.S.; Spear, C.C.; Magnuson, T. Co-regulation of transcription by BRG1 and BRM, two mutually exclusive SWI/SNF ATPase subunits. Epigenet. Chromatin 2017, 10, 62. [Google Scholar] [CrossRef]
- Weng, X.; Yu, L.; Liang, P.; Li, L.; Dai, X.; Zhou, B.; Wu, X.; Xu, H.; Fang, M.; Chen, Q.; et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell Cardiol. 2015, 82, 48–58. [Google Scholar] [CrossRef]
- Tempka, D.; Tokarz, P.; Chmielewska, K.; Kluska, M.; Pietrzak, J.; Rygielska, Z.; Virag, L.; Robaszkiewicz, A. Downregulation of PARP1 transcription by CDK4/6 inhibitors sensitizes human lung cancer cells to anticancer drug-induced death by impairing OGG1-dependent base excision repair. Redox Biol. 2018, 15, 316–326. [Google Scholar] [CrossRef]
- McCabe, M.T.; Davis, J.N.; Day, M.L. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 2005, 65, 3624–3632. [Google Scholar] [CrossRef]
- Sdek, P.; Zhao, P.; Wang, Y.; Huang, C.J.; Ko, C.Y.; Butler, P.C.; Weiss, J.N.; Maclellan, W.R. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J. Cell Biol. 2011, 194, 407–423. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Strober, B.E.; Guha, S.; Khavari, P.A.; Alin, K.; Luban, J.; Begemann, M.; Crabtree, G.R.; Goff, S.P. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 1994, 79, 119–130. [Google Scholar] [CrossRef]
- Sobczak, M.; Pitt, A.R.; Spickett, C.M.; Robaszkiewicz, A. PARP1 Co-Regulates EP300-BRG1-Dependent Transcription of Genes Involved in Breast Cancer Cell Proliferation and DNA Repair. Cancers 2019, 11, 1539. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Transcriptional regulation of human DNA repair genes following genotoxic stress: Trigger mechanisms, inducible responses and genotoxic adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Orlando, K.A.; Nguyen, V.; Raab, J.R.; Walhart, T.; Weissman, B.E. Remodeling the cancer epigenome: Mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev. Anticancer Ther. 2019, 19, 375–391. [Google Scholar] [CrossRef]
- Takao, C.; Morikawa, A.; Ohkubo, H.; Kito, Y.; Saigo, C.; Sakuratani, T.; Futamura, M.; Takeuchi, T.; Yoshida, K. Downregulation of ARID1A, a component of the SWI/SNF chromatin remodeling complex, in breast cancer. J. Cancer 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tolstorukov, M.Y.; Sansam, C.G.; Lu, P.; Koellhoffer, E.C.; Helming, K.C.; Alver, B.H.; Tillman, E.J.; Evans, J.A.; Wilson, B.G.; Park, P.J.; et al. Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc. Natl. Acad. Sci. USA 2013, 110, 10165–10170. [Google Scholar] [CrossRef]
- Wu, Q.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Nickerson, J.A.; Imbalzano, A.N. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics 2017, 9, 919–931. [Google Scholar] [CrossRef]
- Marquez, S.B.; Thompson, K.W.; Lu, L.; Reisman, D. Beyond Mutations: Additional Mechanisms and Implications of SWI/SNF Complex Inactivation. Front. Oncol. 2014, 4, 372. [Google Scholar] [CrossRef]
- Wu, Q.; Madany, P.; Akech, J.; Dobson, J.R.; Douthwright, S.; Browne, G.; Colby, J.L.; Winter, G.E.; Bradner, J.E.; Pratap, J.; et al. The SWI/SNF ATPases Are Required for Triple Negative Breast Cancer Cell Proliferation. J. Cell Physiol. 2015, 230, 2683–2694. [Google Scholar] [CrossRef]
- Do, S.I.; Yoon, G.; Kim, H.S.; Kim, K.; Lee, H.; Do, I.G.; Kim, D.H.; Chae, S.W.; Sohn, J.H. Increased Brahma-related Gene 1 Expression Predicts Distant Metastasis and Shorter Survival in Patients with Invasive Ductal Carcinoma of the Breast. Anticancer Res. 2016, 36, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sharma, S.; Cui, H.; LeBlanc, S.E.; Zhang, H.; Muthuswami, R.; Nickerson, J.A.; Imbalzano, A.N. Targeting the chromatin remodeling enzyme BRG1 increases the efficacy of chemotherapy drugs in breast cancer cells. Oncotarget 2016, 7, 27158–27175. [Google Scholar] [CrossRef] [PubMed]
- Chiba, H.; Muramatsu, M.; Nomoto, A.; Kato, H. Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 1994, 22, 1815–1820. [Google Scholar] [CrossRef]
- DiRenzo, J.; Shang, Y.; Phelan, M.; Sif, S.; Myers, M.; Kingston, R.; Brown, M. BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol. Cell Biol. 2000, 20, 7541–7549. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Ngo, D.; Jacob, J.; Forman, L.W.; Faller, D.V. Prohibitin and the SWI/SNF ATPase subunit BRG1 are required for effective androgen antagonist-mediated transcriptional repression of androgen receptor-regulated genes. Carcinogenesis 2008, 29, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Marshall, T.W.; Link, K.A.; Petre-Draviam, C.E.; Knudsen, K.E. Differential requirement of SWI/SNF for androgen receptor activity. J. Biol. Chem. 2003, 278, 30605–30613. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, J.; Chen, X. The activity of p53 is differentially regulated by Brm- and Brg1-containing SWI/SNF chromatin remodeling complexes. J. Biol. Chem. 2007, 282, 37429–37435. [Google Scholar] [CrossRef]
- Lee, D.; Kim, J.W.; Seo, T.; Hwang, S.G.; Choi, E.J.; Choe, J. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J. Biol. Chem. 2002, 277, 22330–22337. [Google Scholar] [CrossRef]
- Singh, A.P.; Foley, J.F.; Rubino, M.; Boyle, M.C.; Tandon, A.; Shah, R.; Archer, T.K. Brg1 Enables Rapid Growth of the Early Embryo by Suppressing Genes That Regulate Apoptosis and Cell Growth Arrest. Mol. Cell Biol. 2016, 36, 1990–2010. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.R.; Braas, D.; Bhatt, D.M.; Cheng, C.S.; Hong, C.; Doty, K.R.; Black, J.C.; Hoffmann, A.; Carey, M.; Smale, S.T. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 2009, 138, 114–128. [Google Scholar] [CrossRef]
- Wisnik, E.; Ploszaj, T.; Robaszkiewicz, A. Downregulation of PARP1 transcription by promoter-associated E2F4-RBL2-HDAC1-BRM complex contributes to repression of pluripotency stem cell factors in human monocytes. Sci. Rep. 2017, 7, 9483. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.; Nagl, N.G., Jr.; Beck, G.R., Jr.; Moran, E. Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation. J. Biol. Chem. 2009, 284, 10067–10075. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, P.; Ploszaj, T.; Regdon, Z.; Virag, L.; Robaszkiewicz, A. PARP1-LSD1 functional interplay controls transcription of SOD2 that protects human pro-inflammatory macrophages from death under an oxidative condition. Free Radic. Biol. Med. 2019, 131, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A.; Galaxy, T. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobczak, M.; Pietrzak, J.; Płoszaj, T.; Robaszkiewicz, A. BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells. Cancers 2020, 12, 349. https://doi.org/10.3390/cancers12020349
Sobczak M, Pietrzak J, Płoszaj T, Robaszkiewicz A. BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells. Cancers. 2020; 12(2):349. https://doi.org/10.3390/cancers12020349
Chicago/Turabian StyleSobczak, Maciej, Julita Pietrzak, Tomasz Płoszaj, and Agnieszka Robaszkiewicz. 2020. "BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells" Cancers 12, no. 2: 349. https://doi.org/10.3390/cancers12020349
APA StyleSobczak, M., Pietrzak, J., Płoszaj, T., & Robaszkiewicz, A. (2020). BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells. Cancers, 12(2), 349. https://doi.org/10.3390/cancers12020349