The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications




Abstract
:1. Introduction
2. Telomere Maintenance Mechanisms in Human Cancers
3. The Role of Telomere Maintenance Mechanisms in Tumors of Mesenchymal Origin
3.1. The Impact of ALT vs. Telomerase on Clinical Outcome
3.2. Preclinical Evidence of ALT Activation as an Adaptive Response to Telomerase Inhibition
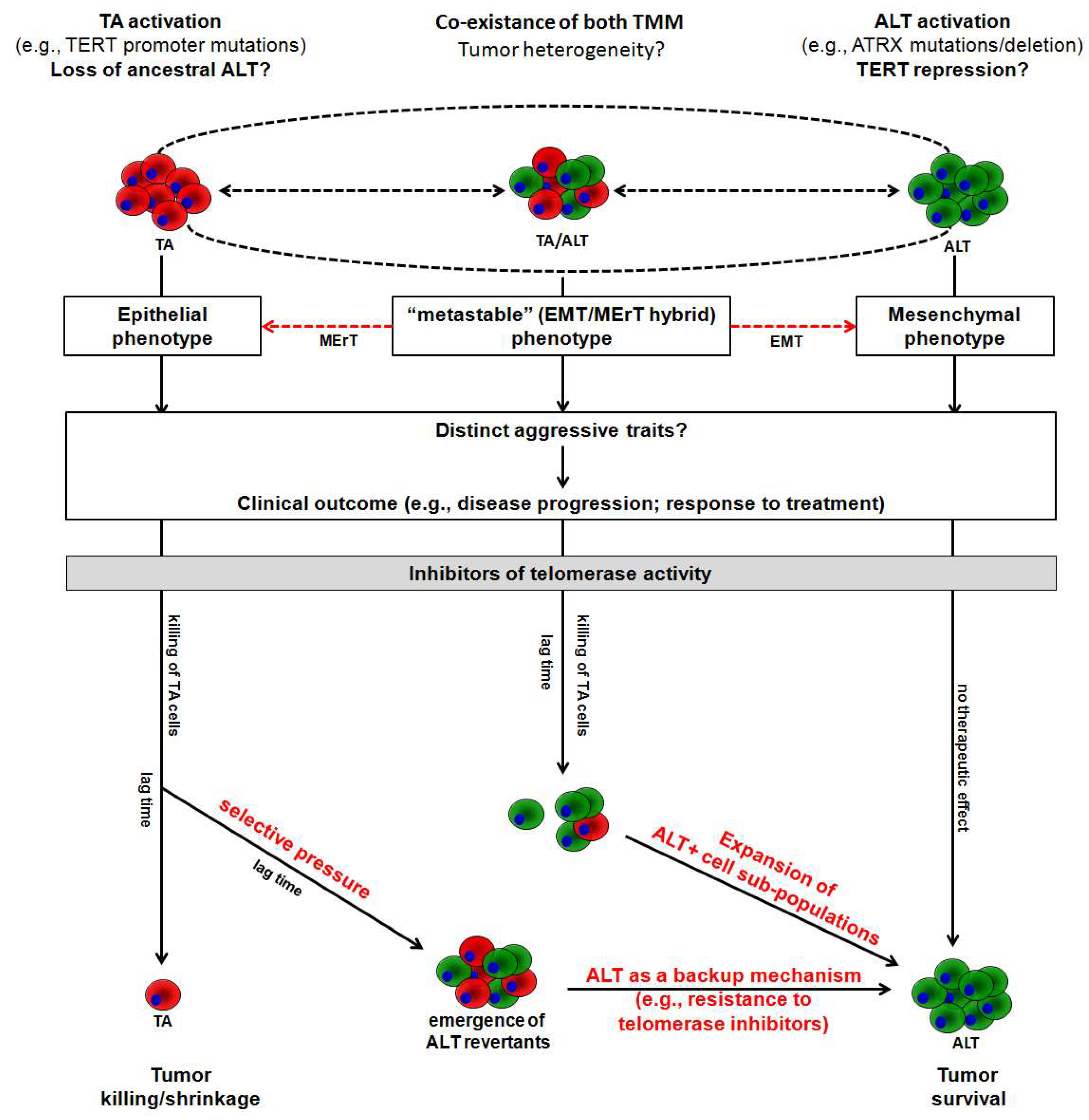
4. Conclusions
Funding
Conflicts of Interest
Appendix A
References
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Turner, K.J.; Vasu, V.; Griffin, D.K. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaswen, P.; MacKenzie, K.L.; Keith, W.N.; Hentosh, P.; Rodier, F.; Zhu, J.; Firestone, G.L.; Matheu, A.; Carnero, A.; Bilsland, A.; et al. Therapeutic targeting of replicative immortality. Semin. Cancer Biol. 2015, 35, S104–S128. [Google Scholar] [CrossRef]
- Gaspar, T.B.; Sá, A.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P.; Vinagre, J. Telomere Maintenance Mechanisms in Cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene 2019. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, E.T.; Zhang, G.; Shay, J.W. In perspective: An update on telomere targeting in cancer. Mol. Carcinog. 2019. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. Febs. Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [Green Version]
- Reddel, R.R. Telomere maintenance mechanisms in cancer: Clinical implications. Curr. Pharm. Des. 2014, 20, 6361–6374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–11855. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Lau, L.M.; Koch, S.; Martin La Rotta, N.; Dagg, R.A.; Reddel, R.R. The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods 2017, 114, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, F.; Gandellini, P.; Cimino-Reale, G.; Zaffaroni, N.; Folini, M. MicroRNA-dependent regulation of telomere maintenance mechanisms: A field as much unexplored as potentially promising. Curr. Pharm. Des. 2014, 20, 6404–6421. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Huang, H.Y.; Otero, J.; Zhao, Z.; Ben-Porat, L.; Satagopan, J.M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Huvos, A.G.; et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res. 2003, 63, 1759–1763. [Google Scholar]
- Sanders, R.P.; Drissi, R.; Billups, C.A.; Daw, N.C.; Valentine, M.B.; Dome, J.S. Telomerase expression predicts unfavorable outcome in osteosarcoma. J. Clin. Oncol. 2004, 22, 3790–3797. [Google Scholar] [CrossRef]
- Li, Y.; Tergaonkar, V. Noncanonical functions of telomerase: Implications in telomerase-targeted cancer therapies. Cancer Res. 2014, 74, 1639–1644. [Google Scholar] [CrossRef] [Green Version]
- Villa, R.; Daidone, M.G.; Motta, R.; Venturini, L.; De Marco, C.; Vannelli, A.; Kusamura, S.; Baratti, D.; Deraco, M.; Costa, A.; et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2008, 14, 4134–4140. [Google Scholar] [CrossRef] [Green Version]
- Venturini, L.; Daidone, M.G.; Motta, R.; Cimino-Reale, G.; Hoare, S.F.; Gronchi, A.; Folini, M.; Keith, W.N.; Zaffaroni, N. Telomere maintenance mechanisms in malignant peripheral nerve sheath tumors: expression and prognostic relevance. Neuro Oncol. 2012, 14, 736–744. [Google Scholar] [CrossRef]
- Liau, J.Y.; Tsai, J.H.; Jeng, Y.M.; Lee, J.C.; Hsu, H.H.; Yang, C.Y. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am. J. Surg. Pathol. 2015, 39, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Park, N.H.; Lee, H. Prognostic value of alternative lengthening of telomeres-associated biomarkers in uterine sarcoma and uterine carcinosarcoma. Int. J. Gynecol. Cancer. 2012, 22, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantù, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere maintenance mechanisms in liposarcomas: Association with histologic subtypes and disease progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Jeng, Y.M.; Liau, J.Y.; Tsai, J.H.; Hsu, H.H.; Yang, C.Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, T.; Shay, J.W.; Wright, W.E.; Hiyama, E.; Shimose, S.; Kubo, T.; Sugita, T.; Yasunaga, Y.; Ochi, M. Telomere-maintenance mechanisms in soft-tissue malignant fibrous histiocytomas. J. Bone Joint Surg. Am. 2009, 91, 928–937. [Google Scholar] [CrossRef]
- Matsuo, T.; Shimose, S.; Kubo, T.; Fujimori, J.; Yasunaga, Y.; Ochi, M. Alternative lengthening of telomeres as a prognostic factor in malignant fibrous histiocytomas of bone. Anticancer Res. 2010, 30, 4959–4962. [Google Scholar]
- Lawlor, R.T.; Veronese, N.; Pea, A.; Nottegar, A.; Smith, L.; Pilati, C.; Demurtas, J.; Fassan, M.; Cheng, L.; Luchini, C. Alternative lengthening of telomeres (ALT) influences survival in soft tissue sarcomas: A systematic review with meta-analysis. BMC Cancer 2019, 19, 232. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Reddel, R.R.; Wright, W.E. Cancer and telomeres—An ALTernative to telomerase. Science 2012, 336, 1388–1390. [Google Scholar] [CrossRef]
- Liau, J.Y.; Lee, J.C.; Tsai, J.H.; Yang, C.Y.; Liu, T.L.; Ke, Z.L.; Hsu, H.H.; Jeng, Y.M. Comprehensive screening of alternative lengthening of telomeres phenotype andloss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Liau, J.Y.; Tsai, J.H.; Yang, C.Y.; Lee, J.C.; Liang, C.W.; Hsu, H.H.; Jeng, Y.M. Alternative lengthening of telomeres phenotype in malignant vascular tumors is highly associated with loss of ATRX expression and is frequently observed in hepatic angiosarcomas. Hum. Pathol. 2015, 46, 1360–1366. [Google Scholar] [CrossRef]
- Slatter, T.L.; Hsia, H.; Samaranayaka, A.; Sykes, P.; Clow, W.B.; Devenish, C.J.; Sutton, T.; Royds, J.A.; Ip, P.P.C.; Cheung, A.N.; et al. Loss of ATRX and DAXX expression identifies poor prognosis for smooth muscle tumours of uncertain malignant potential and early stage uterine leiomyosarcoma. J. Pathol. Clin. Res. 2015, 1, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plantinga, M.J.; Pascarelli, K.M.; Merkel, A.S.; Lazar, A.J.; von Mehren, M.; Lev, D.; Broccoli, D. Telomerase suppresses formation of ALT-associated single-stranded telomeric C-circles. Mol. Cancer Res. 2013, 11, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Gocha, A.R.; Acharya, S.; Groden, J. WRN loss induces switching of telomerase-independent mechanisms of telomere elongation. PLoS ONE 2014, 9, e93991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotillo-Piñeiro, E.; Sierrasesúmaga, L.; Patiñno-García, A. Telomerase activity and telomere length in primary and metastatic tumors from pediatric bone cancer patients. Pediatr. Res. 2004, 55, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Relitti, N.; Saraswati, A.P.; Federico, S.; Khan, T.; Brindisi, M.; Zisterer, D.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Telomerase-based cancer therapeutics: a review on their clinical trials. Curr. Top. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Salloum, R.; Hummel, T.R.; Kumar, S.S.; Dorris, K.; Li, S.; Lin, T.; Daryani, V.M.; Stewart, C.F.; Miles, L.; Poussaint, T.Y.; et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: a pediatric brain tumor consortium study. J. Neurooncol. 2016, 129, 443–451. [Google Scholar] [CrossRef]
- Schrank, Z.; Khan, N.; Osude, C.; Singh, S.; Miller, R.J.; Merrick, C.; Mabel, A.; Kuckovic, A.; Puri, N. Oligonucleotides Targeting Telomeres and Telomerase in Cancer. Molecules 2018, 23, 2267. [Google Scholar] [CrossRef] [Green Version]
- Baerlocher, G.M.; Oppliger Leibundgut, E.; Ottmann, O.G.; Spitzer, G.; Odenike, O.; McDevitt, M.A.; Röth, A.; Daskalakis, M.; Burington, B.; Stuart, M.; et al. Telomerase Inhibitor Imetelstat in Patients with Essential Thrombocythemia. N. Engl. J. Med. 2015, 373, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Begna, K.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.; Mo, Y.; Johnston, J.; Lu, J.; Wientjes, M.G.; Au, J.L. Telomere maintenance in telomerase-positive human ovarian SKOV-3 cells cannot be retarded by complete inhibition of telomerase. FEBS Lett. 2002, 527, 10–14. [Google Scholar] [CrossRef] [Green Version]
- Bechter, O.E.; Zou, Y.; Walker, W.; Wright, W.E.; Shay, J.W. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004, 64, 3444–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queisser, A.; Heeg, S.; Thaler, M.; von Werder, A.; Opitz, O.G. Inhibition of telomerase induces alternative lengthening of telomeres during human esophageal carcinogenesis. Cancer Genet. 2013, 206, 374–386. [Google Scholar] [CrossRef]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, B.K.; Liu, J.P.; Chen, S.M.; Tao, Z.Z. Alternative lengthening of telomeres in hTERT-inhibited laryngeal cancer cells. Cancer Sci. 2010, 101, 1769–1776. [Google Scholar] [CrossRef]
- Ramassone, A.; Pagotto, S.; Veronese, A.; Visone, R. Epigenetics and MicroRNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 459. [Google Scholar] [CrossRef] [Green Version]
- Cimino-Reale, G.; Gandellini, P.; Santambrogio, F.; Recagni, M.; Zaffaroni, N.; Folini, M. miR-380-5p-mediated repression of TEP1 and TSPYL5 interferes with telomerase activity and favours the emergence of an “ALT-like” phenotype in diffuse malignant peritoneal mesothelioma cells. J. Hematol. Oncol. 2017, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Naderlinger, E.; Holzmann, K. Epigenetic Regulation of Telomere Maintenance for Therapeutic Interventions in Gliomas. Genes 2017, 8, 145. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.Q.; Zhong, Z.H.; Henson, J.D.; Reddel, R.R. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 2007, 26, 4635–4647. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Novo, C.L.; Polese, C.; Matheus, N.; Decottignies, A.; Londono-Vallejo, A.; Castronovo, V.; Mottet, D. A new role for histone deacetylase 5 in the maintenance of long telomeres. FASEB J. 2013, 27, 3632–3642. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Gonzalo, S.; Jaco, I.; Muñoz, P.; Gonzalez, S.; Schoeftner, S.; Murchison, E.; Andl, T.; Taiping, C.; Klatt, P.; et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat. Struct. Mol. Biol. 2008, 15, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Kandula, V.; Kosuru, R.; Ye, X.; Irwin, M.G.; Xia, Z. Decoding telomere protein Rap1: Its telomeric and nontelomeric functions and potential implications in diabetic cardiomyopathy. Cell Cycle. 2017, 16, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.M.; Dagg, R.A.; Lau, L.M.S.; Lee, J.H.; Napier, C.E.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.J.; Schiemann, W.P. Means to the ends: The role of telomeres and telomere processing machinery in metastasis. Biochim. Biophys. Acta 2016, 1866, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Sannino, G.; Marchetto, A.; Kirchner, T.; Grünewald, T.G.P. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef] [Green Version]
- Bozzi, F.; Brich, S.; Dagrada, G.P.; Negri, T.; Conca, E.; Cortelazzi, B.; Belfiore, A.; Perrone, F.; Gualeni, A.V.; Gloghini, A.; et al. Epithelioid peritoneal mesothelioma: A hybrid phenotype within a mesenchymal-epithelial/epithelial-mesenchymal transition framework. Oncotarget 2016, 7, 75503–75517. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.R.; Kaul, Z.; Parvin, J.D.; Groden, J. Differential requirements for DNA repair proteins in immortalized cell lines using alternative lengthening of telomere mechanisms. Genes Chromosomes Cancer 2017, 56, 617–631. [Google Scholar] [CrossRef]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Tumor Type | Marker Analyzed | Clinical Endpoint | Outcome | Ref. |
|---|---|---|---|---|
| Osteosarcoma 71 lesions ALT frequency: 66% | TMM 1 | OS | Patients with TMM-negative lesions showed a better outcome (p = 0.05), with 90% (95% CI, 71–100%) 5-year survival compared to 60% (95% CI, 45–76%) for patients whose tumors showed evidence of one or both TMM. | [16] |
| Osteosarcoma 44 lesions ALT frequency 79% | hTERT | DFS; OS | TERT-positive group showed a worse outcome compared to TERT-negative group. Three-year survival estimates were 21.4% ± 9.5% vs. 63.7% ± 11.1% (p = 0.014, DFS) and 42.9% ± 12.2% vs. 70.0 ± 9.9% (p = 0.031, OS), respectively. | [17] |
| TERT/ALT 2 | DFS | A significant difference (p = 0.012) in DFS was observed when the entire group of primary tumors was analyzed according to both TERT and ALT status. Three-year survival estimates were 50.0% ± 17.7% for TERT+/ALT+ patients (n = 6) and 62.3% ± 11.5% for TERT-/ ALT+ patients (n = 29). Patients with TERT+/ALT- lesions (n = 8) experienced disease relapse within 3 years. One patient with a TERT-/ALT- lesion treated with surgery only remained disease-free 13 years after diagnosis. | ||
| TA | DFS | Patients whose tumors had detectable TA (n = 5) experienced an unfavorable outcome compared with patients whose tumors lacked telomerase activity (n = 39). Three-year survival estimates were 20.0% ± 12.6% and 53.4% ± 9.7%, respectively (p = 0.073). | ||
| Diffuse Malignant Peritoneal Mesothelioma 44 lesions ALT frequency 23% | TA/ALT 3 | DFS; DRS | TA proved to be prognostic for the both endpoints (DFS: HR, 3.30; 95% CI, 1.23–8.86, p = 0.018; DRS: HR, 3.56; 95% CI, 1.03–12.5, p = 0.045), whereas ALT failed to significantly affect clinical outcome (DFS: HR, 0.40; 95% CI, 0.14-1.19, p = 0.10; DRS: HR, 0.45; 95% CI, 0.13–1.56; p = 0.21). In a subset of patients (n = 29) with resectable tumors who underwent cytoreductive surgery and hyperthermic intraperitoneal chemotherapy, TA proved to be prognostic for the DFS (HR, 3.32; 95% CI, 1.09–10.12; p = 0.03) and showed a trend towards an association with a poorer DRS (HR, 3.69; 95% CI, 0.79–17.13; p = 0.09). ALT did not affect clinical outcome. | [19] |
| Malignant Peripheral Nerve Sheath Tumors 57 lesions ALT frequency 37% | TA/ALT 4 | DRS | TA proved to be prognostic for the endpoint (HR, 3.78; 95% CI, 0.79–17.13; p = 0.002), even when adjusted for the presence of NF1 syndrome and for margin status after surgical excision. Conversely, ALT status failed to affect clinical outcome, either using APB (HR, 1.25; 95% CI, 0.54–2.89; p = 0.61) or TRF analysis (HR, 0.57; 95% CI, 0.17–1.96; p = 0.38). | [20] |
| Uterine and Retroperitoneal/intra-abdomen Leiomyosarcoma 92 lesions ALT frequency 59% | ALT 5 | OS | In univariate analysis, ALT phenotype was associated with a poor outcome (HR, 2.19; 95% CI, 1.10–4.34; p = 0.025) and showed to be an independent prognostic factor in multivariate analysis (HR, 2.67; 95% CI, 1.08–6.60; p = 0.034). | [21] |
| Uterine sarcoma 41 lesions ALT frequency 46% | ALT 3 | DFS; OS | The presence of APB was a significant prognostic factor for poor DFS (p = 0.018) and OS (p = 0.021). The presence of APBs was not an independent prognostic factor in the multivariate analysis. | [22] |
| Liposarcoma 93 lesions ALT frequency 30% | TMM 1 | DRS | At both univariable and multivariable analysis, TA alone did not prove to be associated with clinical outcome. ALT showed to be a prognostic indicator of unfavorable outcome both at univariable (HR, 2.70; 95% CI, 1.43–5.10; p = 0.0022) and multivariable (adjusted for tumor location, grade, and histology; HR, 3.58; 95% CI, 1.73–7.41; p = 0.0006) analyses. The presence of one or more TMM significantly (HR, 3.73; 95% CI, 1.76–7.88; p = 0.001) affected patient prognosis. Moreover, compared with TMM−cases, increased mortality was observed when TA and ALT phenotypes were considered separately with the TMM+ class, with adjusted hazard ratio estimates from the multivariable model of 2.58 (95% CI, 1.05–6.32; p = 0.0382) and 6.39 (95% CI, 2.64–15.49; p < 0.0001), respectively. | [23] |
| De-differentiated Liposarcoma 46 lesions ALT frequency 30% | ALT 5 | DFS; OS | ALT phenotype was associated with adverse overall survival, albeit not statistically significant (HR, 1.954; p = 0.077). The marker was most significantly correlated with DFS (HR, 3.119; p = 0.003) compared with other clinic-pathological variable, such as mitotic count (HR, 2.689; p = 0.017), grade and stage (HR, 2.689; p = 0.017). | [24] |
| Malignant Fibrous Histiocytomas 43 lesions ALT frequency 33% | TA/ALT 2 | OS | Univariate analysis revealed that ALT (HR, 0.367; 95% CI, 0.135–0.998; p = 0.0495) and TA (HR, 0.403; 95% CI, 0.147–1.107; p = 0.0779) were prognostic risk factors for death, though TA did not reach statistical significance. In the multivariate analysis ALT-positive status was the only independent prognostic factor for death (HR, 0.275; 95% CI, 0.104–0.724; p = 0.0089). | [25] |
| Bone Malignant Fibrous Histiocytomas 10 lesions ALT frequency 50% | TMM 1/TERT | OS | ALT-positive patients had a worse prognosis than other patients (survival rate, 20% vs. 80%, respectively, p = 0.0316) There was no significant correlation between the survival rate and the level of TA (p = 0.923) and of hTERT expression (p = 0.722). | [26] |
| Experimental Model | Telomerase Inhibitor | Outcome | Ref. |
|---|---|---|---|
| Human ovarian cancer cell line | AZT; antisense hTR | maintenance of homogeneous telomere length; no overt features of ALT | [41] |
| Mismatch repair-deficient human colon cancer cells | hTERT dominant negative | high molecular weight TRFs; T-SCE; low tumorigenic potential in nude mice | [42] |
| T-cell lymphomas developing in ATM−/− transgenic mice | Genetic extinction of telomerase | increased heterogeneity in telomere length distribution; increased in APBs; occurrence of extrachromosomal telomere fragments | [43] |
| hTERT-immortalized primary esophageal epithelial cells; transformed human keratinocytes | mutant hTR-expressing lentiviruses; siRNA directed against hTR | heterogeneous telomere length; high frequency of APBs | [44] |
| Fibrosarcoma cell line expressing a deleted form of ACD and RNAi-mediated deletion of ATRX and DAXX | CRISPR/Cas9 knock-out of TERT | increased number of APBs; c-circle DNA production; elongated and heterogeneous telomeres | [45] |
| Human laryngeal cancer cell line | RNAi-mediated depletion of TERT | increased number of APBs; T-SCE | [46] |
| Human diffuse malignant peritoneal mesothelioma cells | miR-380-5p mimic transfection | slightly increased mean telomere length; reduced ATRX expression levels; occurrence of C-circles | [48] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recagni, M.; Bidzinska, J.; Zaffaroni, N.; Folini, M. The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications. Cancers 2020, 12, 949. https://doi.org/10.3390/cancers12040949
Recagni M, Bidzinska J, Zaffaroni N, Folini M. The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications. Cancers. 2020; 12(4):949. https://doi.org/10.3390/cancers12040949
Chicago/Turabian StyleRecagni, Marta, Joanna Bidzinska, Nadia Zaffaroni, and Marco Folini. 2020. "The Role of Alternative Lengthening of Telomeres Mechanism in Cancer: Translational and Therapeutic Implications" Cancers 12, no. 4: 949. https://doi.org/10.3390/cancers12040949