Pediatric Low-Grade Gliomas

Abstract
:1. Introduction
2. Histopathology and Molecular Pathogenesis
2.1. Diffuse Astrocytic Tumors
2.2. Other Astrocytic Tumors
2.3. Neuronal and Mixed Neuronal–Glial Tumors
3. Clinical Presentation
4. Diagnostic Studies
5. Surgical Treatment
5.1. Perioperative Management
5.2. Surgical Planning
6. Adjuvant Therapy, Prognostic Factors, and Outcome
6.1. General Principles for Adjuvant Therapy for Low-Grade Gliomas
6.2. Pilocytic Astrocytoma
6.3. Diffuse Astrocytoma
6.4. Pleomorphic Xanthoastrocytoma
6.5. Subependymal Giant Cell Astrocytoma
6.6. Benign Neuroepithelial Tumors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pollack, I.F. Brain tumors in children. N. Engl. J. Med. 1994, 331, 1500–1507. [Google Scholar] [CrossRef]
- Laws, E.R.J.; Taylor, W.F.; Clifton, M.B.; Okazaki, H. Neurosurgical management of low-grade astrocytoma of the cerebral hemispheres. J. Neurosurg. 1984, 61, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Vertosick, F.T.J.; Selker, R.G.; Arena, V.C. Survival of patients with well-differentiated astrocytomas diagnosed in the era of computed tomography. Neurosurgery 1991, 28, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Meyer, J.; Balss, J.; Capper, D.; Mueller, W.; Christians, A.; Felsberg, J.; Wolter, M.; Mawrin, C.; Wick, W.; et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: A study of 1,010 diffuse gliomas. Acta Neuropathol. 2009, 118, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Pfister, S.M.; Jones, D.T.W. Pediatric gliomas: Current concepts on diagnosis, biology, and clinical management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Hamilton, R.L.; Sobol, R.W.; Nikiforova, M.N.; Lyons-Weiler, M.A.; LaFramboise, W.A.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Holmes, E.J.; et al. Children’s Oncology Group IDH1 mutations are common in malignant gliomas arising in adolescents: A report from the Children’s Oncology Group. Childs Nerv. Syst. 2011, 27, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Lin, A.; Tihan, T.; Burger, P.C.G.E.C. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J. Neuropathol. Exp. Neurol. 2008, 67, 878–887. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, A.; Meyer, J.; Capper, D.; Christians, A.; Remke, M.; Witt, H.; Pfister, S.; von Deimling, A.; Hartmann, C. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009, 118, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Raabe, E.H.; Lim, K.S.; Kim, J.M.; Meeker, A.; Mao, X.; Nikkhah, G.; Maciaczyk, J.; Kahlert, U.; Jain, D.; Bar, E.; et al. BRAF activation induces transformation and then senescence in human neural stem cells: A pilocytic astrocytoma model. Clin. Cancer Res. 2011, 17, 3590–3599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.; Janzarik, W.G.; Remke, M.; Ernst, A.; Werft, W.; Becker, N.; Toedt, G.; Wittmann, A.; Kratz, C.; Olbrich, H.; et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J. Clin. Investig. 2008, 118, 1739–1749. [Google Scholar] [CrossRef]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Ichimura, K.P.C.V. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009, 28, 2119–2123. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Giannini, C.; Scheithauer, B.W. Classification and grading of low-grade astrocytic tumors in children. Brain Pathol. 1997, 7, 785–798. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, G.; Miller, C. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar]
- Ramkissoon, L.A.; Horowitz, P.M.; Craig, J.M.; Ramkissoon, S.H.; Rich, B.E.; Schumacher, S.E.; McKenna, A.; Lawrence, M.S.; Bergthold, G.; Brastianos, P.K.; et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc. Natl. Acad. Sci. USA 2013, 110, 8188–8193. [Google Scholar] [CrossRef] [Green Version]
- Tatevossian, R.G.; Tang, B.; Dalton, J.; Forshew, T.; Lawson, A.R.; Ma, J.; Neale, G.; Al, E. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta Neuropathol. 2010, 120, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Kleihues, P.; Soylemezoglu, F.; Schäuble, B.; Scheithauer, B.W.; Burger, P.C. Histopathology, classification, and grading of gliomas. Glia 1995, 15, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Bouffet, E.; Tabori, U.; Mabbott, D.; Taylor, M.U.B. Rapamycin (sirolimus) in tuberous sclerosis associated pediatric central nervous system tumors. Pediatr. Blood Cancer 2010, 54, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.C.; Lang, F.F.; Epstein, F.J. Central nervous system gangliogliomas: Part 1: Pathology. J. Neurosurg. 1993, 79, 859–866. [Google Scholar] [CrossRef]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef]
- Pekmezci, M.; Villanueva-Meyer, J.E.; Goode, B.; Van Ziffle, J.; Onodera, C.; Grenert, J.P.; Bastian, B.C.; Chamyan, G.; Maher, O.M.; Khatib, Z.; et al. The genetic landscape of ganglioglioma. Acta Neuropathol. Commun. 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Duffner, P.K.; Horowitz, M.E.; Krischer, J.P.; Friedman, H.S.; Burger, P.C.; Cohen, M.E.; Sanford, R.A.; Mulhern, R.K.; James, H.E.; Freeman, C.R. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N. Engl. J. Med. 1993, 328, 1725–1731. [Google Scholar] [CrossRef]
- Wang, A.C.; Jones, D.T.W.; Abecassis, I.J.; Cole, B.L.; Leary, S.E.S.; Lockwood, C.M.; Chavez, L.; Capper, D.; Korshunov, A.; Fallah, A.; et al. Desmoplastic infantile ganglioglioma/astrocytoma (DIG/DIA) are distinct entities with frequent BRAFV600 mutations. Mol. Cancer Res. 2018, 16, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- Rivera, B.; Gayden, T.; Carrot-Zhang, J.; Nadaf, J.; Boshari, T.; Faury, D.; Zeinieh, M.; Blanc, R.; Burk, D.L.; Fahiminiya, S.; et al. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 2016, 131, 847–863. [Google Scholar] [CrossRef]
- Pollack, I.F.; Claassen, D.; Al-Shboul, Q.; Janosky, J.E.; Deutsch, M. Low-grade gliomas of the cerebral hemispheres in children: An analysis of 71 cases. J. Neurosurg. 1995, 82, 536–547. [Google Scholar] [CrossRef]
- Zachenhofer, I.; Donat, M.; Oberndorfer, S.; Roessler, K. Perioperative levetiracetam for prevention of seizures in supratentorial brain tumor surgery. J. Neurooncol. 2011, 101, 101–106. [Google Scholar] [CrossRef]
- Hockey, B.; Leslie, K.; Williams, D. Dexamethasone for intracranial neurosurgery and anaesthesia. J. Clin. Neurosci. 2009, 16, 1389–1393. [Google Scholar] [CrossRef]
- Wisoff, J.H.; Sanford, R.A.; Heier, L.A.; Sposto, R.; Burger, P.C.; Yates, A.J.; Holmes, E.J.; Kun, L.E. Primary neurosurgery for pediatric low-grade gliomas: A prospective multi-institutional study from the Children’s Oncology Group. Neurosurgery 2011, 68, 1548–1555. [Google Scholar] [CrossRef]
- Cohen, K.J.; Pollack, I.F.; Zhou, T.; Buxton, A.; Holmes, E.J.; Burger, P.C.; Brat, D.J.; Rosenblum, M.K.; Hamilton, R.L.; Lavey, R.S.; et al. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro Oncol. 2011, 13, 317–323. [Google Scholar] [CrossRef]
- Finlay, J.L.; Boyett, J.M.; Yates, A.J.; Wisoff, J.H.; Milstein, J.M.; Geyer, J.R.; Bertolone, S.J.; McGuire, P.; Cherlow, J.M.; Tefft, M. Childrens Cancer Group Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. J. Clin. Oncol. 1995, 13, 112–123. [Google Scholar] [CrossRef]
- Hirsch, J.F.; Sainte Rose, C.; Pierre-Kahn, A.; Pfister, A.; Hoppe-Hirsch, E. Benign astrocytic and oligodendrocytic tumors of the cerebral hemispheres in children. J. Neurosurg. 1989, 70, 568–572. [Google Scholar] [CrossRef]
- Schneider, W.; Noll, D.C.; Cohen, J.D. Functional topographic mapping of the cortical ribbon in human vision with conventional MRI scanners. Nature 1993, 365, 150–153. [Google Scholar] [CrossRef]
- Moshel, Y.A.; Elliott, R.E.; Monoky, D.J.; Wisoff, J.H. Role of diffusion tensor imaging in resection of thalamic juvenile pilocytic astrocytoma. J. Neurosurg. Pediatr. 2009, 4, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.S.; Kincaid, J.; Ojemann, G.A.; Lettich, E. Brain mapping techniques to maximize resection, safety, and seizure control in children with brain tumors. Neurosurgery 1989, 25, 786–792. [Google Scholar] [CrossRef]
- Packer, R.J.; Sutton, L.N.; Patel, K.M.; Duhaime, A.C.; Schiff, S.; Weinstein, S.R.; Gaillard, W.D.; Conry, J.A.; Schut, L. Seizure control following tumor surgery for childhood cortical low-grade gliomas. J. Neurosurg. 1994, 80, 998–1003. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.S.; Ghatan, S.; Haglund, M.M.; Dobbins, J.; Ojemann, G.A. Low-grade gliomas associated with intractable epilepsy: Seizure outcome utilizing electrocorticography during tumor resection. J. Neurosurg. 1993, 79, 62–69. [Google Scholar] [CrossRef]
- Iannelli, A.; Guzzetta, F.; Battaglia, D.; Iuvone, L.; Di Rocco, C. Surgical treatment of temporal tumors associated with epilepsy in children. Pediatr. Neurosurg. 2000, 32, 248–254. [Google Scholar] [CrossRef]
- Palma, L.; Guidetti, B. Cystic pilocytic astrocytomas of the cerebral hemispheres: Surgical experience with 51 cases and long-term results. J. Neurosurg. 1985, 62, 811–815. [Google Scholar] [CrossRef]
- Broniscer, A.; Baker, S.J.; West, A.N.; Fraser, M.M.; Proko, E.; Kocak, M.; Dalton, J.; Zambetti, G.P.; Ellison, D.W.; Kun, L.E.; et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J. Clin. Oncol. 2007, 25, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Ater, J.L.; Zhou, T.; Holmes, E.; Mazewski, C.M.; Booth, T.N.; Freyer, D.R.; Lazarus, K.H.; Packer, R.J.; Prados, M.; Sposto, R.; et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 2641–2647. [Google Scholar] [CrossRef] [Green Version]
- Fisher, P.G.; Tihan, T.; Goldthwaite, P.T.; Wharam, M.D.; Carson, B.S.; Weingart, J.D.; Repka, M.X.; Cohen, K.J.; Burger, P.C. Outcome analysis of childhood low-grade astrocytomas. Pediatr. Blood Cancer 2008, 51, 245–250. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bowers, D.C.; Krause, T.P.; Aronson, L.J.; Barzi, A.; Burger, P.C.; Carson, B.S.; Weingart, J.D.; Wharam, M.D.; Melhem, E.R.; Cohen, K.J. Second surgery for recurrent pilocytic astrocytoma in children. Pediatr. Neurosurg. 2001, 34, 229–234. [Google Scholar] [CrossRef]
- Philippon, J.H.; Clemenceau, S.H.; Fauchon, F.H.; Foncin, J.F. Supratentorial low-grade astrocytomas in adults. Neurosurgery 1993, 32, 554–559. [Google Scholar] [CrossRef]
- Hanzély, Z.; Polgár, C.; Fodor, J.; Brucher, J.M.; Vitanovics, D.; Mangel, L.C.; Áfra, D. Role of early radiotherapy in the treatment of supratentorial WHO Grade II astrocytomas: Long-term results of 97 patients. J. Neurooncol. 2003, 63, 305–312. [Google Scholar] [CrossRef]
- Dirks, P.B.; Jay, V.; Becker, L.E.; Drake, J.M.; Humphreys, R.P.; Hoffman, H.J.; Rutka, J.T. Development of anaplastic changes in low-grade astrocytomas of childhood. Neurosurgery 1994, 34, 68–78. [Google Scholar]
- Chadderton, R.D.; West, C.G.; Schuller, S.; Quirke, D.C.; Gattamaneni, R.; Taylor, R. Radiotherapy in the treatment of low-grade astrocytomas. Childs Nerv. Syst. 1995, 11, 443–448. [Google Scholar] [CrossRef]
- Ellenberg, L.; McComb, J.G.; Siegel, S.E.; Stowe, S. Factors affecting intellectual outcome in pediatric brain tumor patients. Neurosurgery 1987, 21, 638–644. [Google Scholar] [CrossRef]
- Livesey, E.A.; Hindmarsh, P.C.; Brook, C.G.; Whitton, A.C.; Bloom, H.J.G.; Tobias, J.S.; Godlee, J.N.; Britton, J. Endocrine disorders following treatment of childhood brain tumours. Br. J. Cancer 1990, 61, 622–625. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Liu, J.-F.; Tatevossian, R.G.; Chiang, J.; Qaddoumi, I.; Gajjar, A.; Walker, D.; Harreld, J.; Merchant, T.E.; Ellison, D.W. Risk stratification in pediatric low-grade glioma and glioneuronal tumor treated with radiation therapy: An integrated clinicopathologic and molecular analysis. Neuro. Oncol. 2020, 61, 622–625. [Google Scholar] [CrossRef]
- Cherlow, J.M.; Shaw, D.W.W.; Margraf, L.R.; Bowers, D.C.; Huang, J.; Fouladi, M.; Onar-Thomas, A.; Zhou, T.; Pollack, I.F.; Gajjar, A.; et al. Conformal radiation therapy for pediatric patients with low-grade glioma: Results from the children’s oncology group phase 2 study ACNS0221. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Merchant, T.E.; Kun, L.E.; Wu, S.; Xiong, X.; Sanford, R.A.; Boop, F.A. Phase II trial of conformal radiation therapy for pediatric low-grade glioma. J. Clin. Oncol. 2009, 27, 3598–3604. [Google Scholar] [CrossRef] [Green Version]
- Breneman, J.C.; Donaldson, S.S.; Constine, L.; Merchant, T.; Marcus, K.; Paulino, A.C.; Followill, D.; Mahajan, A.; Laack, N.; Esiashvili, N.; et al. The children’s oncology group radiation oncology discipline: 15 years of contributions to the treatment of childhood cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 860–874. [Google Scholar] [CrossRef]
- Kano, H.; Niranjan, A.; Kondziolka, D.; Flickinger, J.C.; Pollack, I.F.; Jakacki, R.I.; Lunsford, L.D. Stereotactic radiosurgery for pilocytic astrocytomas: Part 2: Outcomes in pediatric patients. J. Neurooncol. 2009, 95, 219–229. [Google Scholar] [CrossRef]
- Boëthius, J.; Ulfarsson, E.; Rähn, T.; Lippittz, B. Gamma knife radiosurgery for pilocytic astrocytomas. J. Neurosurg. 2002, 97, 677–680. [Google Scholar] [CrossRef]
- Kreth, F.W.; Faist, M.; Warnke, P.C.; Rossner, R.; Volk, B.; Ostertag, C.B. Interstitial radiosurgery of low-grade gliomas. J. Neurosurg. 1995, 82, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.J.; Goumnerova, L.; Billett, A.L.; Lavally, B.; Scott, R.M.; Bishop, K.; Xu, R.; Poussaint, T.Y.; Kieran, M.; Kooy, H.; et al. Stereotactic radiotherapy for localized low-grade gliomas in children: Final results of a prospective trial. Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.K.; Squire, S.; Lamborn, K.; Banerjee, A.; Gupta, N.; Wara, W.M.; Prados, M.D.; Berger, M.S.; Haas-Kogan, D.A. Phase II TPDCV protocol for pediatric low-grade hypothalamic/chiasmatic gliomas: 15-year update. J. Neurooncol. 2010, 100, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Gnekow, A.K.; Walker, D.A.; Kandels, D.; Picton, S.; Giorgio, P.; Grill, J.; Stokland, T.; Sandstrom, P.E.; Warmuth-Metz, M.; Pietsch, T.; et al. A European randomised controlled trial of the addition of etoposide to standard vincristine and carboplatin induction as part of an 18-month treatment programme for childhood (</=16 years) low grade glioma—A final report. Eur. J. Cancer 2017, 81, 206–225. [Google Scholar] [PubMed] [Green Version]
- Lassaletta, A.; Scheinemann, K.; Zelcer, S.M.; Hukin, J.; Wilson, B.A.; Jabado, N.; Carret, A.S.; Lafay-Cousin, L.; Larouche, V.; Hawkins, C.E.; et al. Phase II weekly vinblastine for chemotherapy-naive children with progressive low-grade glioma: A canadian pediatric brain tumor consortium study. J. Clin. Oncol. 2016, 34, 3537–3543. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, C.; Di Patre, P.L.; Schüler, D.; Schüler, G.; Yaşargil, M.G.; Yonekawa, Y.; Lütolf, U.M.; Kleihues, P.; Ohgaki, H. A population-based study of the incidence and survival rates in patients with pilocytic astrocytoma. J. Neurosurg. 2003, 98, 1170–1174. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol. 2017, 19, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Fangusaro, J.; Onar-Thomas, A.; Poussaint, T.Y.; Wu, S.; Ligon, A.H.; Lindeman, N.; Others, A. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Gururangan, S.; Fangusaro, J.; Poussaint, T.Y.; McLendon, R.E.; Onar-Thomas, A.; Wu, S.; Packer, R.J.; Banerjee, A.; Gilbertson, R.J.; Fahey, F.; et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas—A Pediatric Brain Tumor Consortium study. Neuro Oncol. 2014, 16, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Sievert, A.J.; Lang, S.-S.; Boucher, K.L.; Madsen, P.J.; Slaunwhite, E.; Choudhari, N.; Kellet, M.; Storm, P.B.; Resnick, A.C. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc. Natl. Acad. Sci. USA 2013, 110, 5957–5962. [Google Scholar] [CrossRef] [Green Version]
- Bid, H.K.; Kibler, A.; Phelps, D.A.; Manap, S.; Xiao, L.; Lin, J.; Capper, D.; Oswald, D.; Geier, B.; DeWire, M.; et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin. Cancer Res. 2013, 19, 6716–6729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorani, I. Genetically engineered mouse models of gliomas: Technological developments for translational discoveries. Cancers 2019, 11, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, C.; Walker, E.; Mohamed, N.; Zhang, C.; Jacob, K.; Shirinian, M.; Alon, N.; Kahn, D.; Fried, I.; Scheinemann, K.; et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin. Cancer Res. 2011, 17, 4790–4798. [Google Scholar] [CrossRef] [Green Version]
- Babu, R.; Bagley, J.H.; Park, J.G.; Friedman, A.H.; Adamson, C. Low-grade astrocytomas: The prognostic value of fibrillary, gemistocytic, and protoplasmic tumor histology. J. Neurosurg. 2013, 119, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Pahapill, P.A.; Ramsay, D.A.; Del Maestro, R.F. Pleomorphic xanthoastrocytoma: Case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery 1996, 38, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Jenkins, J.; Burger, P.; Langston, J.; Merchant, T.; Heideman, R.; Thompson, S.; Sanford, A.; Kun, L.; Gajjar, A. Pleomorphic xanthoastrocytoma: Favorable outcome after complete surgical resection. Neuro Oncol. 2001, 3, 184–192. [Google Scholar] [CrossRef]
- Macaulay, R.J.; Jay, V.; Hoffman, H.J.; Becker, L.E. Increased mitotic activity as a negative prognostic indicator in pleomorphic xanthoastrocytoma. Case report. J. Neurosurg. 1993, 79, 761–768. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Shigemori, M.; Okamoto, K.; Morimatsu, M.; Arakawa, M.; Nakayama, K. Clinicopathological study of pleomorphic xanthoastrocytoma: Correlation between histological features and prognosis. Pathol. Int. 2000, 50, 703–708. [Google Scholar] [CrossRef]
- Cuccia, V.; Zuccaro, G.; Sosa, F.; Monges, J.; Lubienieky, F.; Taratuto, A.L. Subependymal giant cell astrocytoma in children with tuberous sclerosis. Childs Nerv. Syst. 2003, 19, 232–243. [Google Scholar] [CrossRef]
- Sinson, G.; Sutton, L.N.; Yachnis, A.T.; Duhaime, A.C.; Schut, L. Subependymal giant cell astrocytomas in children. Pediatr. Neurosurg. 1994, 20, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Care, M.M.; Agricola, K.; Tudor, C.; Mays, M.; Franz, D.N. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology 2013, 80, 574–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, S.F.; Moore, S.A.; Menezes, A.H.; VanGilder, J.C. Ganglioglioma: 13 years of experience. Neurosurgery 1992, 31, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Tamiya, T.; Ono, Y.; Furuta, T.; Asari, S.; Ohmoto, T. Cerebral gangliogliomas: Clinical characteristics, CT and MRI. Acta Neurochir. 1999, 141, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Taratuto, A.L.; Pomata, H.; Sevlever, G.; Gallo, G.; Monges, J. Dysembryoplastic neuroepithelial tumor: Morphological, immunocytochemical, and deoxyribonucleic acid analyses in a pediatric series. Neurosurgery 1995, 36, 474–481. [Google Scholar] [CrossRef]
- Sukheeja, D.; Mehta, J. Dysembryoplastic neuroepithelial tumor: A rare brain tumor not to be misdiagnosed. Asian J. Neurosurg 2016, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Pilcher, W.H.; Silbergeld, D.L.; Berger, M.S.; Ojemann, G.A. Intraoperative electrocorticography during tumor resection: Impact on seizure outcome in patients with gangliogliomas. J. Neurosurg. 1993, 78, 891–902. [Google Scholar] [CrossRef]
- Sugiyama, K.; Arita, K.; Shima, T.; Nakaoka, M.; Matsuoka, T.; Taniguchi, E.; Okamura, T.; Yamasaki, H.; Kajiwara, Y.; Kurisu, K. Good clinical course in infants with desmoplastic cerebral neuroepithelial tumor treated by surgery alone. J. Neurooncol. 2002, 59, 63–69. [Google Scholar] [CrossRef]
- Mallucci, C.; Lellouch-Tubiana, A.; Salazar, C.; Cinalli, G.; Renier, D.; Sainte-Rose, C.; Pierre-Kahn, A.; Zerah, M. The management of desmoplastic neuroepithelial tumours in childhood. Childs Nerv. Syst. 2000, 16, 8–14. [Google Scholar] [CrossRef]
- Duffner, P.K.; Burger, P.C.; Cohen, M.E.; Sanford, R.A.; Krischer, J.P.; Elterman, R.; Aronin, P.A.; Pullen, J.; Horowitz, M.E.; Parent, A.; et al. Desmoplastic infantile gangliogliomas: An approach to therapy. Neurosurgery 1994, 34, 583–589. [Google Scholar] [CrossRef]
- Bächli, H.; Avoledo, P.; Gratzl, O.; Tolnay, M. Therapeutic strategies and management of desmoplastic infantile ganglioglioma: Two case reports and literature overview. Childs Nerv. Syst. 2003, 19, 359–366. [Google Scholar] [PubMed] [Green Version]
- Tamburrini, G.; Colosimo, C., Jr.; Giangaspero, F.; Riccardi, R.; Di Rocco, C. Desmoplastic infantile ganglioglioma. Childs Nerv. Syst. 2003, 19, 292–297. [Google Scholar] [CrossRef] [PubMed]



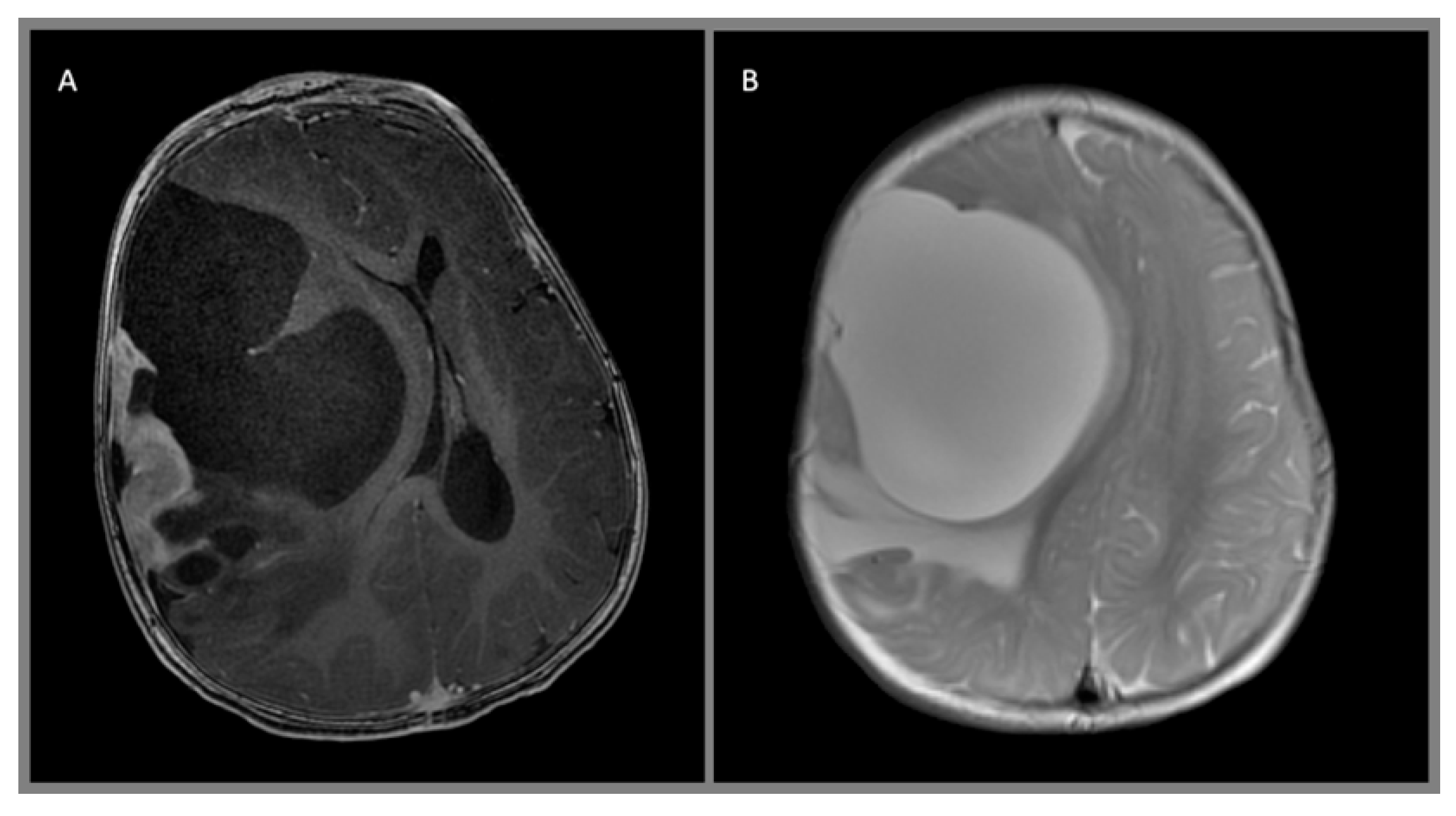


{kind=link}
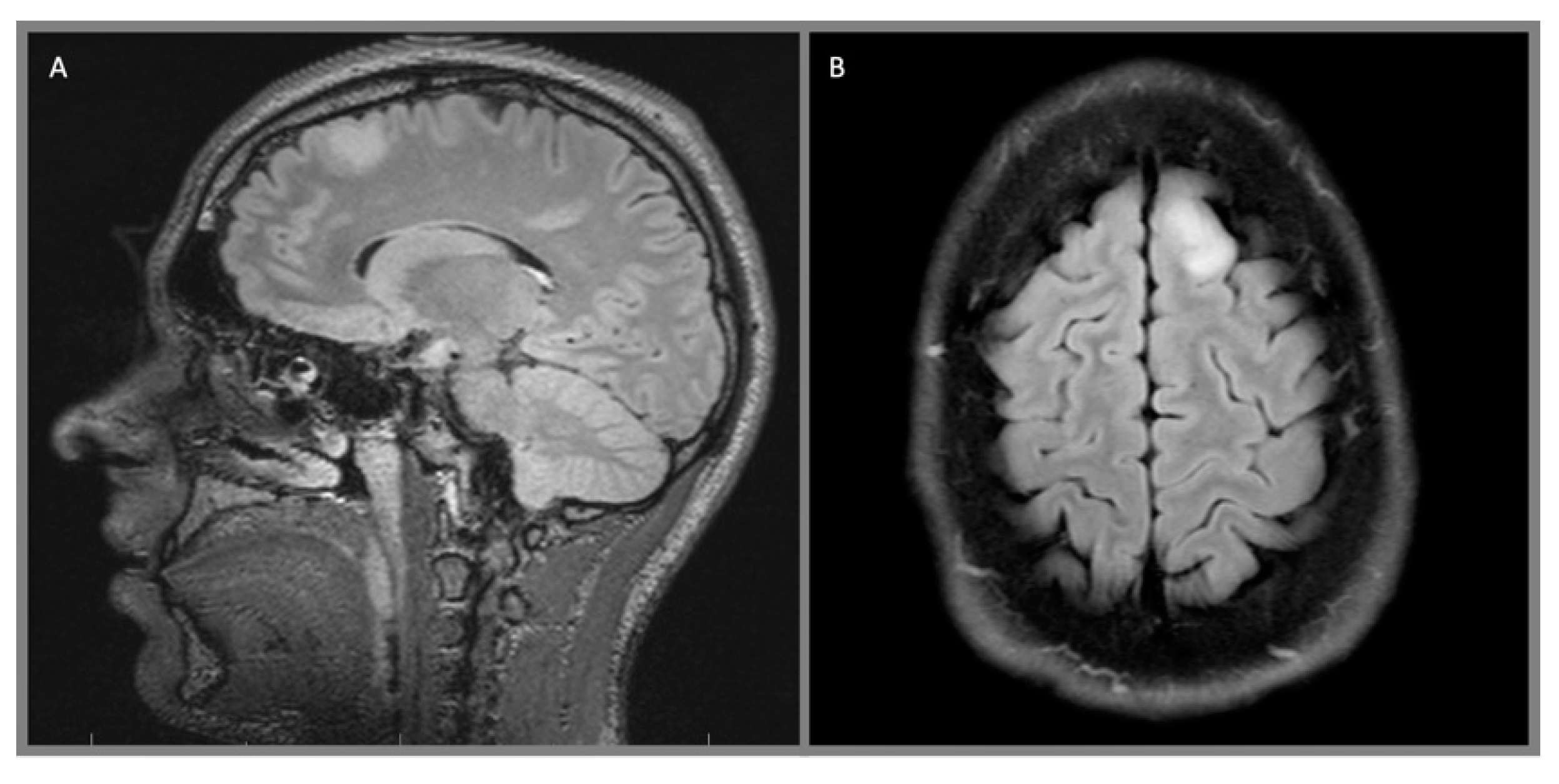
{kind=link}
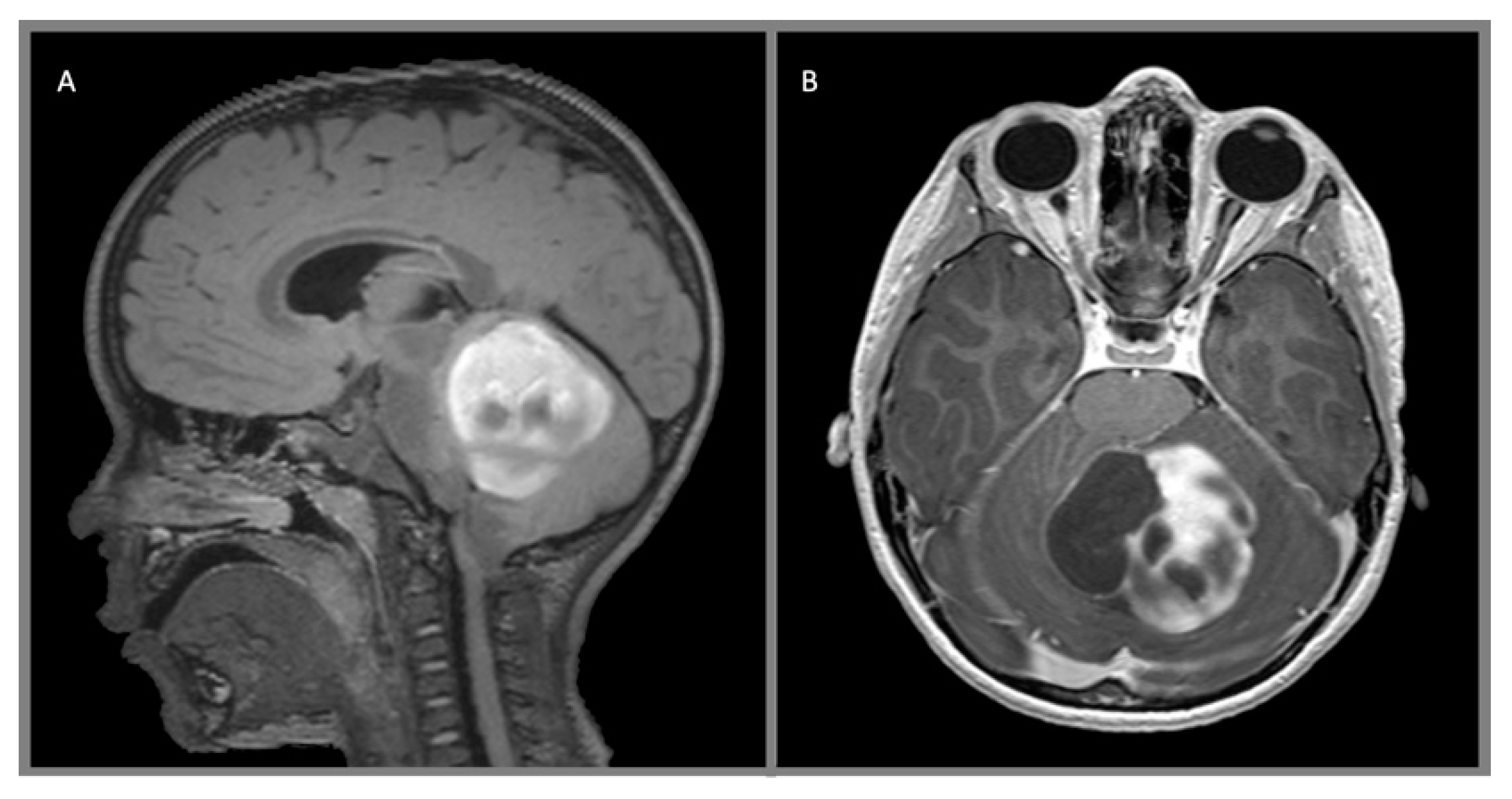
{kind=link}
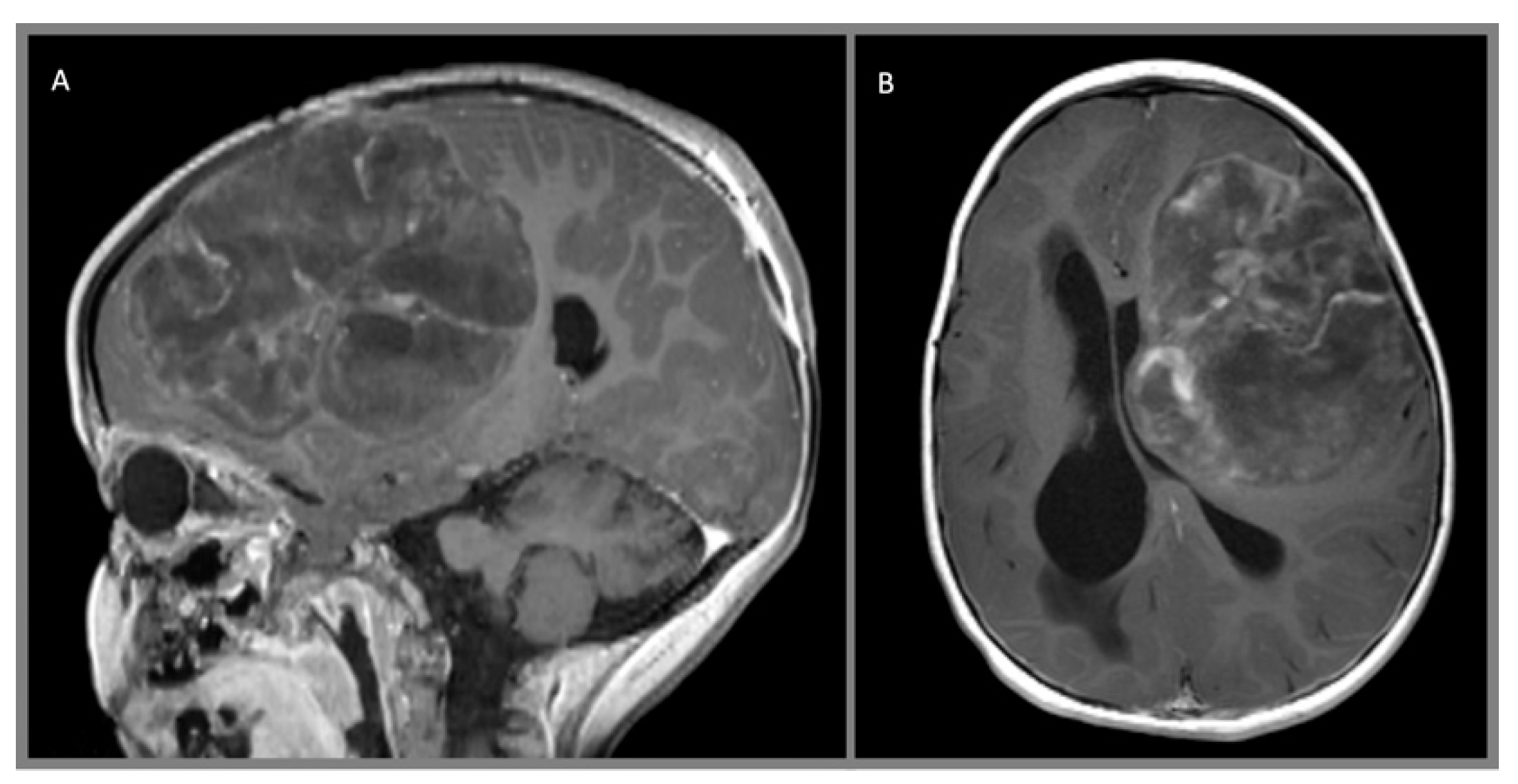
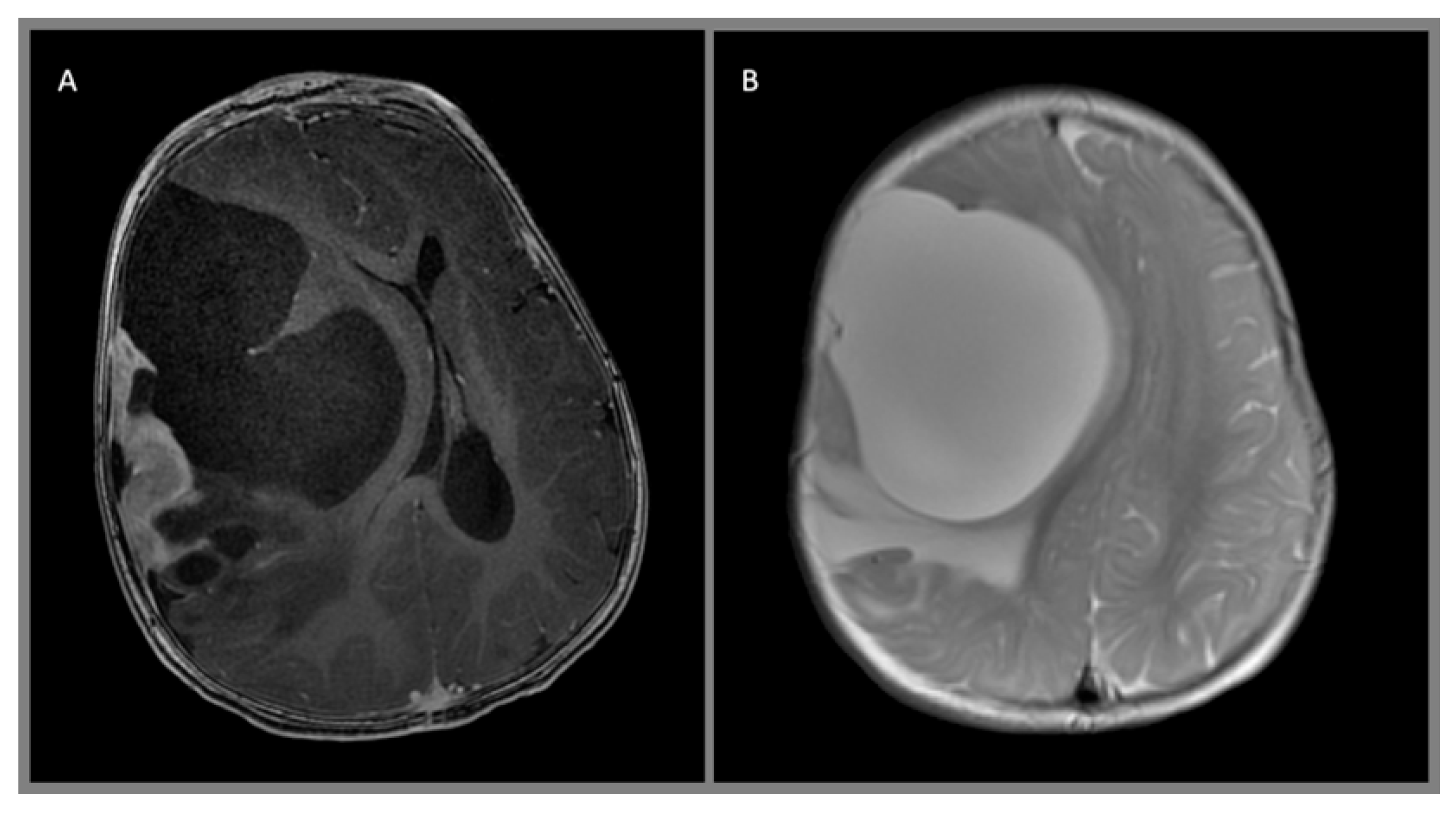{kind=link}
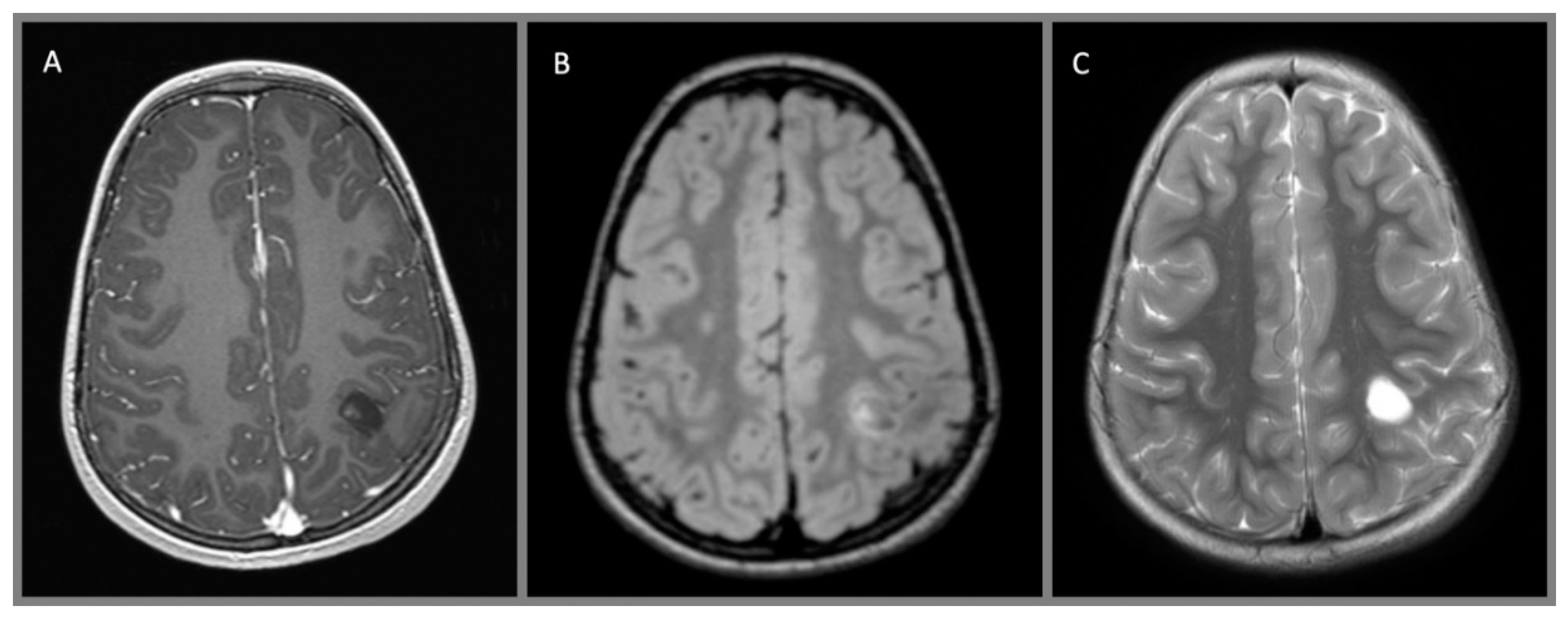{kind=link}
| Tumor Class | Tumor Type | WHO Grade |
|---|---|---|
| Diffuse Astrocytic and Oligodendroglial Tumors | Diffuse astrocytoma | II |
| Oligodendroglioma | II | |
| Other Astrocytic Tumors | Pilocytic astrocytoma | I |
| Subependymal giant cell astrocytoma | I | |
| Pleomorphic xanthoastrocytoma | II | |
| Ependymal Tumors | Subependymoma | I |
| Myxopapillary ependymoma | I | |
| Ependymoma | II | |
| Other Gliomas | Angiocentric glioma | I |
| Chordoid glioma of the third ventricle | II | |
| Neuronal and Mixed Neuronal–Glial Tumors | Dysembryoplastic neuroepithelial tumor | I |
| Gangliocytoma | I | |
| Ganglioglioma | I | |
| Dysplastic gangliocytoma of cerebellum (Lhermitte–Duclos) | I | |
| Desmoplastic infantile astrocytoma and ganglioglioma | I | |
| Papillary glioneuronal tumor | I | |
| Rosette-forming glioneuronal tumor | II |
| Tumor | Histology | Molecular Features | Management | Outcomes |
|---|---|---|---|---|
| Pilocytic astrocytoma | Compact bipolar astrocytes with long GFAP-positive processes, eosinophilic granular bodies, Rosenthal fibers, microcysts, leptomeningeal infiltration, glomeroid vascular proliferation, and mitoses. | A total of 80% exhibit BRAF gene or other MAPK signaling pathway alterations (BRAFV600E, BRAF/KIAA1549 translocation, neurofibromin mutation, etc.). | Gross total resection (GTR) is the surgical goal. Biologic agents such as selumetinib, vemurafenib, dabrafenib and trametinib, as well as traditional chemotherapy and radiotherapy are treatment options for unresectable residual or recurrent disease. | After GTR, the five-year PFS is 75%–100%. Subtotally resected tumors have a five-year PFS of approximately 50%–80%. |
| Diffuse astrocytoma | Diffuse infiltration of well-differentiated neoplastic astrocytes. Mitotic activity is absent. | Amplification and/or rearrangement of MYB/MYBL1 [18,19,20]. | GTR can be curative. Subtotal or no resection may be treated with vincristine plus carboplatin or vinblastine monotherapy [45]. | Five-year PFS of 55% and OS of 87%. Reviewed diagnosis shows three-year PFS of 40% and five-year OS of 48% [46]. |
| Pleomorphic xanthoastrocytoma | Dense cellularity and nuclear atypia with pleomorphism and multinucleation, low mitotic index, lipid-rich “xanthomatous” astrocytes, extracellular reticulin, eosinophilic granular bodies, and lymphocytic infiltrate. | BRAFV600E mutations and 9p21 (CDKN2A/B) deletions may be seen [9]. | GTR is the goal. BRAFV600E -targeted therapy such as vemurafenib/dabrafenib or other MAP kinase pathway-targeted therapy may be possible. Adjuvant therapy is utilized only for tumors which progress and are thought to be unresectable. | GTR results in 90% long-term survival at five years and 80% at ten years, versus 65% at five years for incompletely resected tumors. |
| Subependymal giant cell astrocytoma | Large gemistocytic, spindled, and ganglion-cell like astrocytes. Immunoreactivity for both glial and neuronal markers is often observed. Perivascular pseudopalisading may be seen, mitoses are not. | Dysregulation of mTOR signaling linked with tuberous sclerosis. Germline mutations in TSC1 or TSC2 in up to 20% of patients [47]. | GTR is the goal. Molecular therapy targeting dysregulated mTOR signaling such as everolimus/sirolimus and radiotherapy or stereotactic radiosurgery are used for unresectable recurrence. | Total or near total resection results in an excellent prognosis. Subtotally resected lesions tend to enlarge over time. |
| Ganglioglioma | Highly differentiated binucleated ganglion cells in a background of astrocytes or oligodendrocytes. | BRAF alterations, particularly BRAFV600E, or downstream members of the MAP kinase pathway [25,26]. A small subset exhibit CDKN2A deletion [26]. | GTR is the goal. BRAFV600E -targeted therapy such as vemurafenib/dabrafenib or other MAP kinase pathway-targeted therapy may be possible. Adjuvant therapy is utilized only for tumors which progress and are thought to be unresectable. | Five-year survival rate exceeding 90%. |
| Dysembryoplastic neuroepithelial tumor | Nodules of oligodendroglial-like cells and/or focal cortical dysplasia intermixed with a looser textured component containing “floating neurons” in a mucinous matrix. | FGFR1 alterations and MAP kinase pathway activation are frequent [29]. | Surgical excision often curative. MAP kinase pathway-targeted therapy may be possible. Adjuvant therapy is utilized only for tumors which progress and are thought to be unresectable. | Favorable outcome, particularly after GTR. While seizure control after resection was studied, we are unaware of any large series evaluating survival outcome. |
| Desmoplastic infantile ganglioglioma | Dense, fibrous, desmoplastic stroma containing a mixture of neuroepithelial cells with both astrocytic and neuronal differentiation. | Frequently have BRAFV600E mutations [28]. | Surgical resection can be curative. The treatment of any residual tumor is controversial as spontaneous regression can occur. | Rare enough that no large series to evaluate outcome is known to the authors. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, K.L.; Pollack, I.F. Pediatric Low-Grade Gliomas. Cancers 2020, 12, 1152. https://doi.org/10.3390/cancers12051152
Collins KL, Pollack IF. Pediatric Low-Grade Gliomas. Cancers. 2020; 12(5):1152. https://doi.org/10.3390/cancers12051152
Chicago/Turabian StyleCollins, Kelly L., and Ian F. Pollack. 2020. "Pediatric Low-Grade Gliomas" Cancers 12, no. 5: 1152. https://doi.org/10.3390/cancers12051152
APA StyleCollins, K. L., & Pollack, I. F. (2020). Pediatric Low-Grade Gliomas. Cancers, 12(5), 1152. https://doi.org/10.3390/cancers12051152