Transcriptional Regulation of CCL2 by PARP1 Is a Driver for Invasiveness in Breast Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PARP1 Inhibition Negatively Affect Breast Cancer Cell Proliferation and Migration
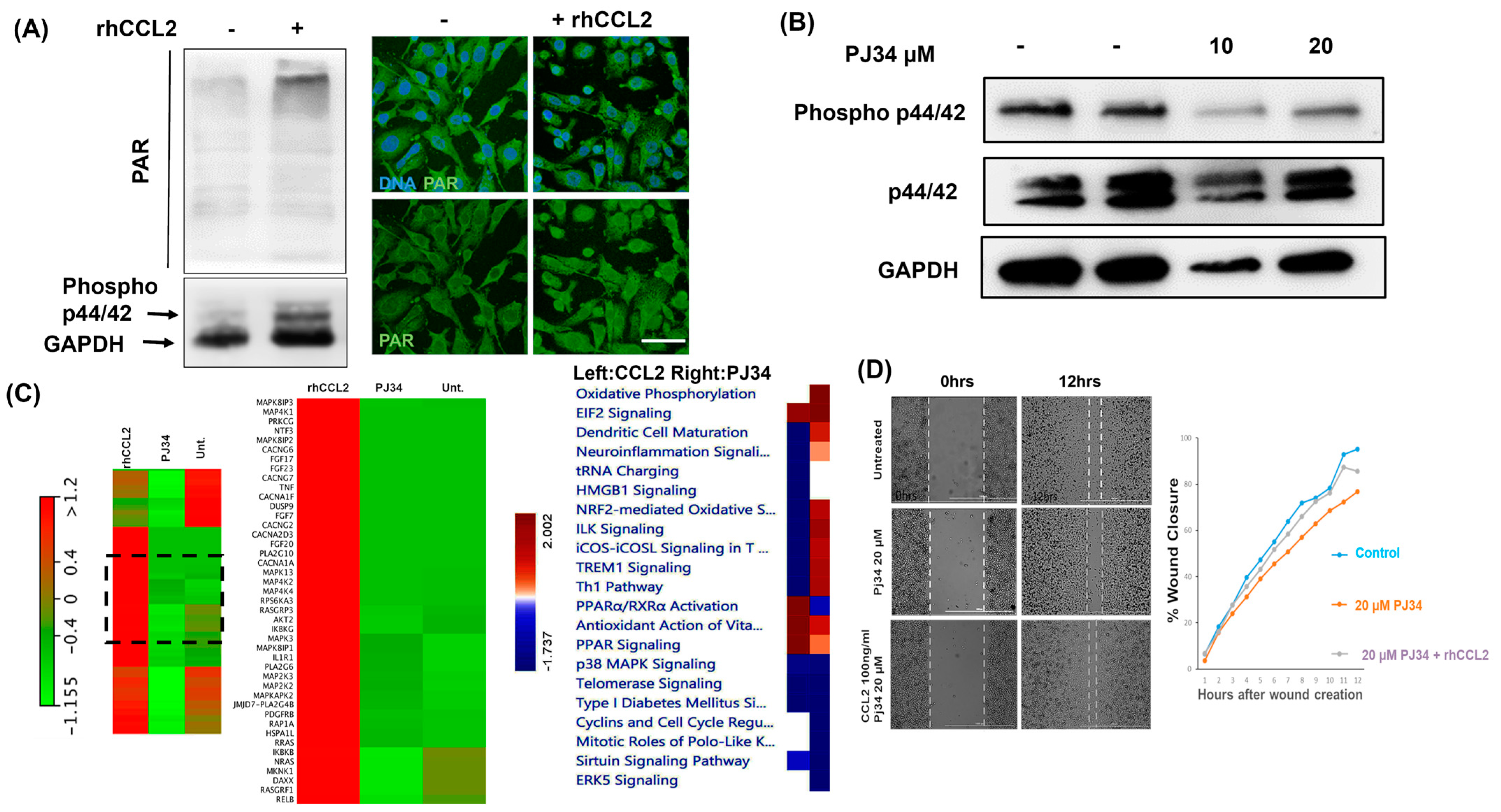
2.2. PARP1 Inhibition Affects Cytokine-Mediated Cellular Signaling in Breast Cancer Cells
2.3. PARP1 Transcriptionally Regulates CCL2 in Breast Cancer
2.4. PAR Level Is Associated with CCL2 Transcription in Breast Cancer
2.5. PARP1 and NFκB Interaction is Essential for CCL2 Transcription in Breast Cancer
2.6. Crosstalk between PARP1 and CCL2 Regulates Invasiveness in Breast Cancer
2.7. CCL2 and NFκB Correlate in Breast Cancer Patients Affecting Relapsed Free Survival
3. Discussion
4. Materials and Methods
4.1. Cell Cultures, Antibodies, and Reagents
4.2. Whole Transcriptome Analysis
4.3. Enzyme-Linked Immunosorbent Assay (ELISA) for Human CCL2
4.4. RNA. Isolation and Quantitative Real-Time PCR
4.5. Cell Proliferation and Colony Formation Assay
4.6. Wound Healing and Boyden Chamber Invasion Assay
4.7. Western Blotting
4.8. Transfection of shRNA/siRNA and Generation of Stable Lines with shRNA Knockdown
4.9. Co-Immunoprecipitation and Chromatin Immunoprecipitation
4.10. Gel Shift Assay with NFκB Probes
4.11. Immunofluorescence with MB-231 Cells
4.12. Annexin V Apoptosis Assay
4.13. Statistics and Analysis of Publicly Available Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ali, S.; Lazennec, G. Chemokines: Novel targets for breast cancer metastasis. Cancer Metastasis Rev. 2007, 26, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast cancer tumor stroma: Cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications, and therapeutic opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.; Sarkissyan, M.; Wu, Y.; Vadgama, J.V. Groalpha overexpression drives cell migration and invasion in triple-negative breast cancer cells. Oncol. Rep. 2017, 38, 21–30. [Google Scholar] [CrossRef]
- Dutta, P.; Sarkissyan, M.; Paico, K.; Wu, Y.; Vadgama, J.V. Mcp-1 is overexpressed in triple-negative breast cancers and drives cancer invasiveness and metastasis. Breast Cancer Res. Treat. 2018, 170, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. Parp inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef]
- Donizy, P.; Pietrzyk, G.; Halon, A.; Kozyra, C.; Gansukh, T.; Lage, H.; Surowiak, P.; Matkowski, R. Nuclear-cytoplasmic parp-1 expression as an unfavorable prognostic marker in lymph node-negative early breast cancer: 15-year follow-up. Oncol. Rep. 2014, 31, 1777–1787. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of parp1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, Y.; Wang, L.; Luo, X.; Huang, K.; Wang, C.; Du, M.; Liu, F.; Luo, T.; Huang, D.; et al. Poly(adp-ribose) polymerase 1 is a key regulator of estrogen receptor alpha-dependent gene transcription. J. Biol. Chem. 2013, 288, 11348–11357. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a parp-1 and nuclear factor-{kappa}b-associated secretome (pnas). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Abdelkarim, G.E.; Gertz, K.; Harms, C.; Katchanov, J.; Dirnagl, U.; Szabo, C.; Endres, M. Protective effects of pj34, a novel, potent inhibitor of poly(adp-ribose) polymerase (parp) in in vitro and in vivo models of stroke. Int. J. Mol. Med. 2001, 7, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, R.; Pirnes-Karhu, S.; Pietiläinen, K.H.; Pirinen, E. Adipose tissue nad(+)-homeostasis, sirtuins and poly(adp-ribose) polymerases -important players in mitochondrial metabolism and metabolic health. Redox Biol. 2017, 12, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. Pgc-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Burkle, A.; Virag, L. Poly(adp-ribose): Paradigms and paradoxes. Mol. Asp. Med. 2013, 34, 1046–1065. [Google Scholar] [CrossRef]
- James, D.I.; Smith, K.M.; Jordan, A.M.; Fairweather, E.E.; Griffiths, L.A.; Hamilton, N.S.; Hitchin, J.R.; Hutton, C.P.; Jones, S.; Kelly, P.; et al. First-in-class chemical probes against poly(adp-ribose) glycohydrolase (parg) inhibit DNA repair with differential pharmacology to olaparib. ACS Chem. Biol. 2016, 11, 3179–3190. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor nf-kappa B pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Martin-Oliva, D.; O’Valle, F.; Munoz-Gamez, J.A.; Valenzuela, M.T.; Nunez, M.I.; Aguilar, M.; de Ruiz Almodovar, J.M.; Garcia del Moral, R.; Oliver, F.J. Crosstalk between parp-1 and nf-kappa B modulates the promotion of skin neoplasia. Oncogene 2004, 23, 5275–5283. [Google Scholar] [CrossRef]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced ikappa B alpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of parp-1 is not required for the nf-kappa b coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent parp-1 activation by phosphorylated erk2 increases elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Visochek, L.; Cohen-Armon, M. Parp1-erk synergism in proliferating cells. Oncotarget 2018, 9, 29140–29145. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2010, 15, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Shou, Q.; Fu, H.; Huang, X.; Yang, Y. Parp-1 controls NK cell recruitment to the site of viral infection. JCI Insight 2019, 4, e121291. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.I.; Ha, H.C. Parp1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain Behav. 2014, 4, 552–565. [Google Scholar] [CrossRef]
- Nalabothula, N.; Al-jumaily, T.; Eteleeb, A.M.; Flight, R.M.; Xiaorong, S.; Moseley, H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Genome-wide profiling of parp1 reveals an interplay with gene regulatory regions and DNA methylation. PLoS ONE 2015, 10, e0135410. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the ccl2-ccr2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef]
- Ke, Y.; Han, Y.; Guo, X.; Wen, J.; Wang, K.; Jiang, X.; Tian, X.; Ba, X.; Boldogh, I.; Zeng, X. Parp1 promotes gene expression at the post-transcriptional level by modulating the RNA-binding protein hur. Nat. Commun. 2017, 8, 14632. [Google Scholar] [CrossRef]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Wahner Hendrickson, A.E.; Menefee, M.E.; Hartmann, L.C.; Long, H.J.; Northfelt, D.W.; Reid, J.M.; Boakye-Agyeman, F.; Kayode, O.; Flatten, K.S.; Harrell, M.I.; et al. A phase I clinical trial of the poly(adp-ribose) polymerase inhibitor veliparib and weekly topotecan in patients with solid tumors. Clin. Cancer Res. 2018, 24, 744. [Google Scholar] [CrossRef]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.E.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Overall survival in a trial of orally administered ccr2 inhibitor ccx872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J. Clin. Oncol. 2018, 36, 92. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Dis. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. BioRxiv 2019, 326470. [Google Scholar] [CrossRef]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, P.; Paico, K.; Gomez, G.; Wu, Y.; Vadgama, J.V. Transcriptional Regulation of CCL2 by PARP1 Is a Driver for Invasiveness in Breast Cancer. Cancers 2020, 12, 1317. https://doi.org/10.3390/cancers12051317
Dutta P, Paico K, Gomez G, Wu Y, Vadgama JV. Transcriptional Regulation of CCL2 by PARP1 Is a Driver for Invasiveness in Breast Cancer. Cancers. 2020; 12(5):1317. https://doi.org/10.3390/cancers12051317
Chicago/Turabian StyleDutta, Pranabananda, Kimberly Paico, Gabriela Gomez, Yanyuan Wu, and Jaydutt V. Vadgama. 2020. "Transcriptional Regulation of CCL2 by PARP1 Is a Driver for Invasiveness in Breast Cancer" Cancers 12, no. 5: 1317. https://doi.org/10.3390/cancers12051317