ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Patient Samples
2.2. MTT Cell Viability and BrdU Cell Proliferation Assay
2.3. Transfection and Cell Line Selection
2.4. Systemic Leukemia Animal Model
2.5. Bioluminescence (IVIS) Imaging
2.6. Flow Cytometry Analysis
2.7. Real-Time Quantitative Polymerase Chain Reaction (Q-PCR)
2.8. Absolute Q-PCR
2.9. Protein Extraction, Western Blotting, And Antibodies
2.10. Plasmid Construction and Lentiviral Production
2.11. Design of on-Target and off-Target sgRNAs for the ABL and mABL Gene
2.12. Sanger Sequencing and Gene Editing Efficiency Assay
2.13. RNA-Guided Engineered Nuclease-Restriction Fragment Length Polymorphism (RGEN-RFLP) Assay
2.14. Karyotype and Fluorescent In-Situ Hybridization (FISH) Analysis
2.15. T-Cell Development and Lineage In Vivo
2.16. Flow Cytometric Analysis and Cell Staining
2.17. Statistical Methods
2.18. Ethics approval and consent to participate
3. Results
3.1. Optimization of Viral Transduction for Human K562 Cells
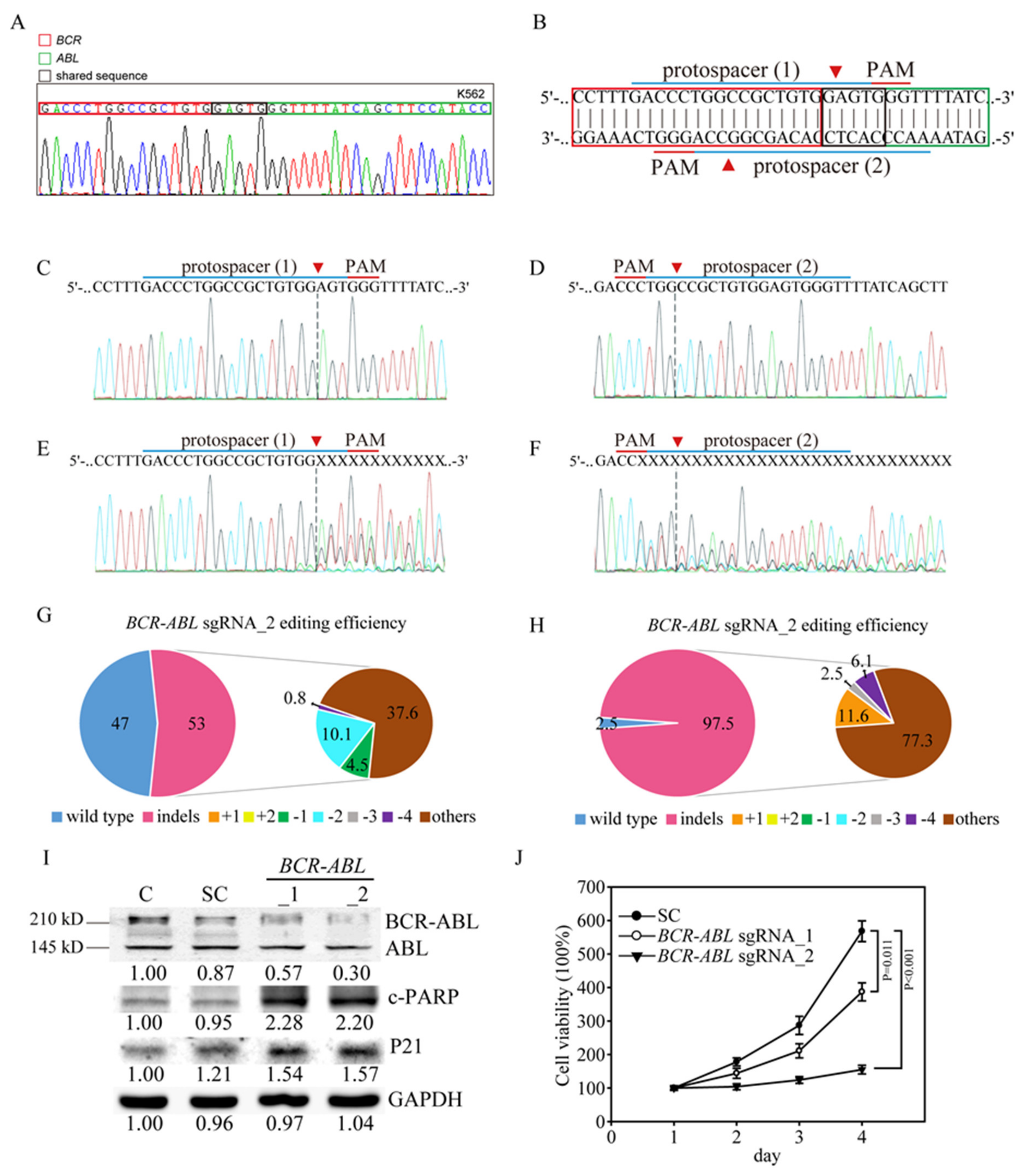
3.2. Efficient and Specific CRISPR/Cas9 Gene Editing of the Human BCR-ABL Junctions in K562 CML Cells
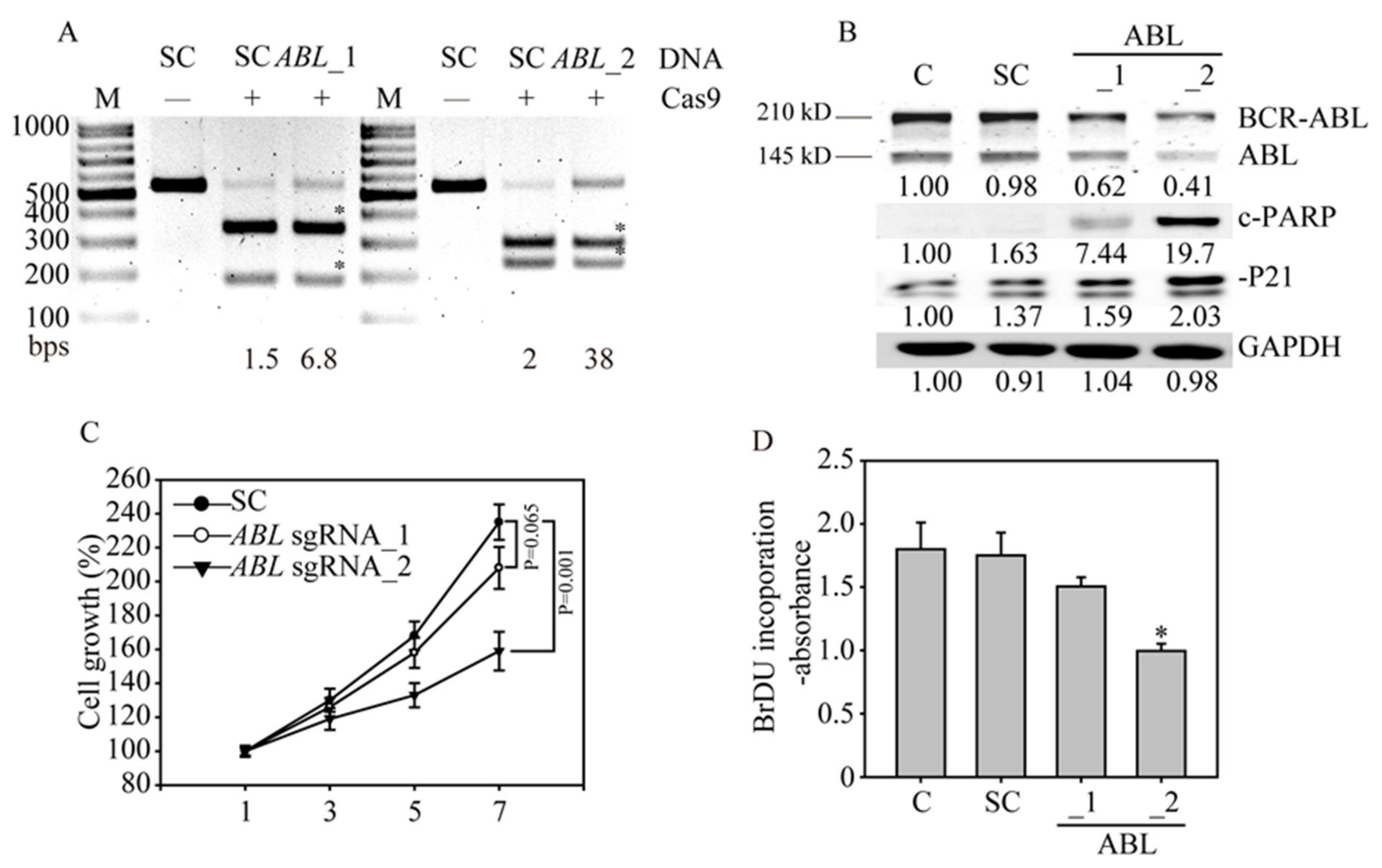
3.3. CRISPR/Cas9 Sufficiently Disrupts the ABL Gene in K562 Cells
3.4. ABL Loss Significantly Inhibits K562 Cancer Cell Growth and Induces Apoptosis
3.5. In Vivo ABL-Targeted Gene Editing Effectively Inhibits Leukemia Cell Growth
3.6. Ex Vivo ABL-Targeted Gene Editing of Clinical CML Patients
3.7. High Specificity of the ABL-Targeted CRISPR/Cas9 System Without Potential off-Target Effects
3.8. The ABL Gene Editing Showed Better Anticancer Effects than Imatinib Treatment in Imatinib-Resistant K562 Cells
3.9. Mouse ABL Editing through CRISPR/Cas9 to Investigate T-Cell Survival and Development In Vivo
3.10. Summary of the Research Strategy in this Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| Ph | Philadelphia chromosome |
| TKIs | Tyrosine kinase inhibitors |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| RGEN-RFLP | RNA-guided engineered nuclease-restriction fragment length polymorphism |
| TIDE | Tracking of indels by decomposition |
| SCID | Severe combined immunodeficiency |
| IVIS | In vivo imaging system |
References
- Roy, M.; Sarkar, R.; Mukherjee, A.; Mukherjee, S. Inhibition of crosstalk between Bcr-Abl and PKC signaling by PEITC, augments imatinib sensitivity in chronic myelogenous leukemia cells. Chem. Interact. 2015, 242, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Castagnetti, F.; Gugliotta, G.; Breccia, M.; Iurlo, A.; Levato, L.; Albano, F.; Vigneri, P.G.; Abruzzese, E.; Rossi, G.; Rupoli, S.; et al. The BCR-ABL1 transcript type influences response and outcome in Philadelphia chromosome-positive chronic myeloid leukemia patients treated frontline with imatinib. Am. J. Hematol. 2017, 92, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Nashed, A.L.; Rao, K.W.; Gulley, M.L. Clinical Applications of BCR-ABL Molecular Testing in Acute Leukemia. J. Mol. Diagn. 2003, 5, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K. Tumour promoting functions of TGF-β in CML-initiating cells. J. Biochem. 2012, 152, 383–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Rowley, J.D. Chronic Myeloid Leukemia: Current Perspectives. Clin. Lab. Med. 2011, 31, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, S.; Ph.D., H.Z. Induction of Chronic Myeloid Leukemia in Mice. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2016; Volume 1465, pp. 17–25. [Google Scholar]
- Burchert, A.; Saussele, S.; Eigendorff, E.; Müller, M.C.; Sohlbach, K.; Inselmann, S.; Schütz, C.; Metzelder, S.K.; Ziermann, J.; Kostrewa, P.; et al. Interferon alpha 2 maintenance therapy may enable high rates of treatment discontinuation in chronic myeloid leukemia. Leukemiak 2015, 29, 1331–1335. [Google Scholar] [CrossRef]
- Heim, D. Tyrosinkinase-Inhibitoren in der Behandlung der chronisch myeloischen Leukämie. Ther. Umsch. 2006, 63, 249–254. [Google Scholar] [CrossRef]
- Campbell, T.; Felsten, L.; Moore, J. Disappearance of Lentigines in a Patient Receiving Imatinib Treatment for Familial Gastrointestinal Stromal Tumor Syndrome. Arch. Dermatol. 2009, 145, 1313–1316. [Google Scholar] [CrossRef] [Green Version]
- Bhamidipati, P.K.; Kantarjian, H.; Cortes, J.; Cornelison, A.M.; Jabbour, E.J. Management of imatinib-resistant patients with chronic myeloid leukemia. Ther. Adv. Hematol. 2012, 4, 103–117. [Google Scholar] [CrossRef]
- Frank, O.; Brors, B.; Fabarius, A.; Li, L.; Haak, M.; Merk, S.; Schwindel, U.; Zheng, C.; Müller, M.C.; Gretz, N.; et al. Gene expression signature of primary imatinib-resistant chronic myeloid leukemia patients. Leukemiak 2006, 20, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Mou, H.; Kennedy, Z.; Anderson, D.G.; Yin, H.; Xue, W. Precision cancer mouse models through genome editing with CRISPR-Cas9. Genome Med. 2015, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jínek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wang, P.; Parton, T.; Zhou, Y.; Chrysovergis, K.; Rockowitz, S.; Chen, W.-Y.; Abdel-Wahab, O.; Wade, P.A.; Zheng, D.; et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016, 30, 92–107. [Google Scholar] [CrossRef] [PubMed]
- García-Tuñón, I.; Hernández-Sánchez, M.; Ordoñez, J.L.; Alonso-Pérez, V.; Álamo-Quijada, M.; Benito, R.; Guerrero, C.; Hernández-Rivas, J.M.; Sánchez-Martín, M. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitro and in a xenograft model of chronic myeloid leukemia. Oncotarget 2017, 8, 26027–26040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; A Bartley, P.; Slader, C.; Field, C.; Dang, P.; Filshie, R.J.; et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia 2010, 24, 1719–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-H.; Huang, C.-S.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chang, Y.-J.; Tam, K.-W.; Wei, P.-L.; Cheng, T.-C.; Chu, J.-S.; et al. Overexpression and Activation of the α9-Nicotinic Receptor During Tumorigenesis in Human Breast Epithelial Cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.-H.; Hsieh, Y.; Huang, L.-C.; Lin, C.-Y.; Hsu, K.-W.; Hsieh, W.-S.; Chi, W.-M.; Lee, C.-H. A rapid and quantitative method to detect human circulating tumor cells in a preclinical animal model. BMC Cancer 2017, 17, 440. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2013, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Bennett, M.J.; Luistro, L.; Carvajal, D.; Nevins, T.; Smith, M.; Tyagi, G.; Cai, J.; Wei, X.; Lin, T.-A.; et al. Discovery of siRNA Lipid Nanoparticles to Transfect Suspension Leukemia Cells and Provide In Vivo Delivery Capability. Mol. Ther. 2013, 22, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Chasseriau, J.; Rivet, J.; Bilan, F.; Chomel, J.-C.; Guilhot, F.; Bourmeyster, N.; Kitzis, A. Characterization of the Different BCR-ABL Transcripts with a Single Multiplex RT-PCR. J. Mol. Diagn. 2004, 6, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Malhotra, A.; Dutta, A. Detection of DNA fusion junctions for BCR-ABL translocations by Anchored ChromPET. Genome Med. 2010, 2, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- VanDyke, K.; Fitter, S.; Drew, J.; Fukumoto, S.; Schultz, C.G.; Sims, N.; Yeung, D.T.; Hughes, T.P.; Zannettino, A.C. Prospective Histomorphometric and DXA Evaluation of Bone Remodeling in Imatinib-Treated CML Patients: Evidence for Site-Specific Skeletal Effects. J. Clin. Endocrinol. Metab. 2013, 98, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-H.; Chow, J.-M.; Hsieh, Y.-Y.; Lin, C.-Y.; Hsu, K.-W.; Hsieh, W.-S.; Chi, W.-M.; Shabangu, B.; Lee, C.-H. HDAC1,2 Knock-Out and HDACi Induced Cell Apoptosis in Imatinib-Resistant K562 Cells. Int. J. Mol. Sci. 2019, 20, 2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberman, I.; Sionov, R.V.; Zuckerman, V.; Haupt, S.; Goldberg, Z.; Strasser, A.; Ben-Sasson, Z.S.; Baniyash, M.; Koleske, A.J.; Haupt, Y. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle 2008, 7, 3847–3857. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Zhang, N.; He, Y.-W.; Koleske, A.J.; Pendergast, A.M. Defective T cell development and function in the absence of Abelson kinases. J. Immunol. 2007, 179, 7334–7343. [Google Scholar] [CrossRef] [Green Version]
- Ginn, S.L.; Edelstein, M.L.; Abedi, M.R.; Hamlin, N.; Alexander, I.E.; Wixon, J. Gene therapy clinical trials worldwide to 2012 – an update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef]
- Stone, D.; Niyonzima, N.; Jerome, K.R. Genome editing and the next generation of antiviral therapy. Qual. Life Res. 2016, 135, 1071–1082. [Google Scholar] [CrossRef] [Green Version]
- Yee, J.-K. Off-target effects of engineered nucleases. FEBS J. 2016, 283, 3239–3248. [Google Scholar] [CrossRef]
- Lekomtsev, S.; Aligianni, S.; Lapao, A.; Buerckstuemmer, T. Efficient generation and reversion of chromosomal translocations using CRISPR/Cas technology. BMC Genom. 2016, 17, 739. [Google Scholar] [CrossRef] [Green Version]
- Hara, S.; Tamano, M.; Yamashita, S.; Kato, T.; Saito, T.; Sakuma, T.; Yamamoto, T.; Inui, M.; Takada, S. Generation of mutant mice via the CRISPR/Cas9 system using FokI-dCas9. Sci. Rep. 2015, 5, 11221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Héon, E.; Hauswirth, W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, J.V. 2 BCR-ABL gene variants. Baillière’s Clin. Haematol. 1997, 10, 203–222. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | SC (n = 4) | mABL sgRNA_1 (n = 3) | mABL sgRNA_2 n = 3 |
|---|---|---|---|
| RBC (×106/uL) | 9.72 ± 0.49 | 10.04 ± 0.3 | 10.04 ± 0.29 |
| WBC (×103/ uL) | 3.77 ± 1.12 | 6.42 ± 1.06 | 4.24 ± 0.5 |
| Hemoglobin (g/dL) | 14.63 ± 0.79 | 15.17 ± 0.38 | 15.1 ± 0.42 |
| Hematocrit (%) | 45.83 ± 2.72 | 48.6 ± 1.4 | 47.13 ± 1.1 |
| Neutrophils (%) | 14.43 ± 1.22 | 13.07 ± 1.47 | 22.97 ± 4.8 |
| Lymphocytes (%) | 72.33 ± 4.76 | 82.4 ± 1.8 | 70.37 ± 6.96 |
| Monocytes (U/Ul) | 11.45 ± 5.56 | 10.1 ± 1.01 | 9.47 ± 1.91 |
| Platelets (x103/uL) | 563.25 ± 130.27 | 521.67 ± 90.24 | 599.33 ± 129.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-H.; Hsieh, Y.-Y.; Tzeng, H.-E.; Lin, C.-Y.; Hsu, K.-W.; Chiang, Y.-S.; Lin, S.-M.; Su, M.-J.; Hsieh, W.-S.; Lee, C.-H. ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo. Cancers 2020, 12, 1399. https://doi.org/10.3390/cancers12061399
Chen S-H, Hsieh Y-Y, Tzeng H-E, Lin C-Y, Hsu K-W, Chiang Y-S, Lin S-M, Su M-J, Hsieh W-S, Lee C-H. ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo. Cancers. 2020; 12(6):1399. https://doi.org/10.3390/cancers12061399
Chicago/Turabian StyleChen, Shu-Huey, Yao-Yu Hsieh, Huey-En Tzeng, Chun-Yu Lin, Kai-Wen Hsu, Yun-Shan Chiang, Su-Mei Lin, Ming-Jang Su, Wen-Shyang Hsieh, and Chia-Hwa Lee. 2020. "ABL Genomic Editing Sufficiently Abolishes Oncogenesis of Human Chronic Myeloid Leukemia Cells In Vitro and In Vivo" Cancers 12, no. 6: 1399. https://doi.org/10.3390/cancers12061399