Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Characterization of the Cohort and Identification of FGFR1 Amplified Breast Cancer Cases
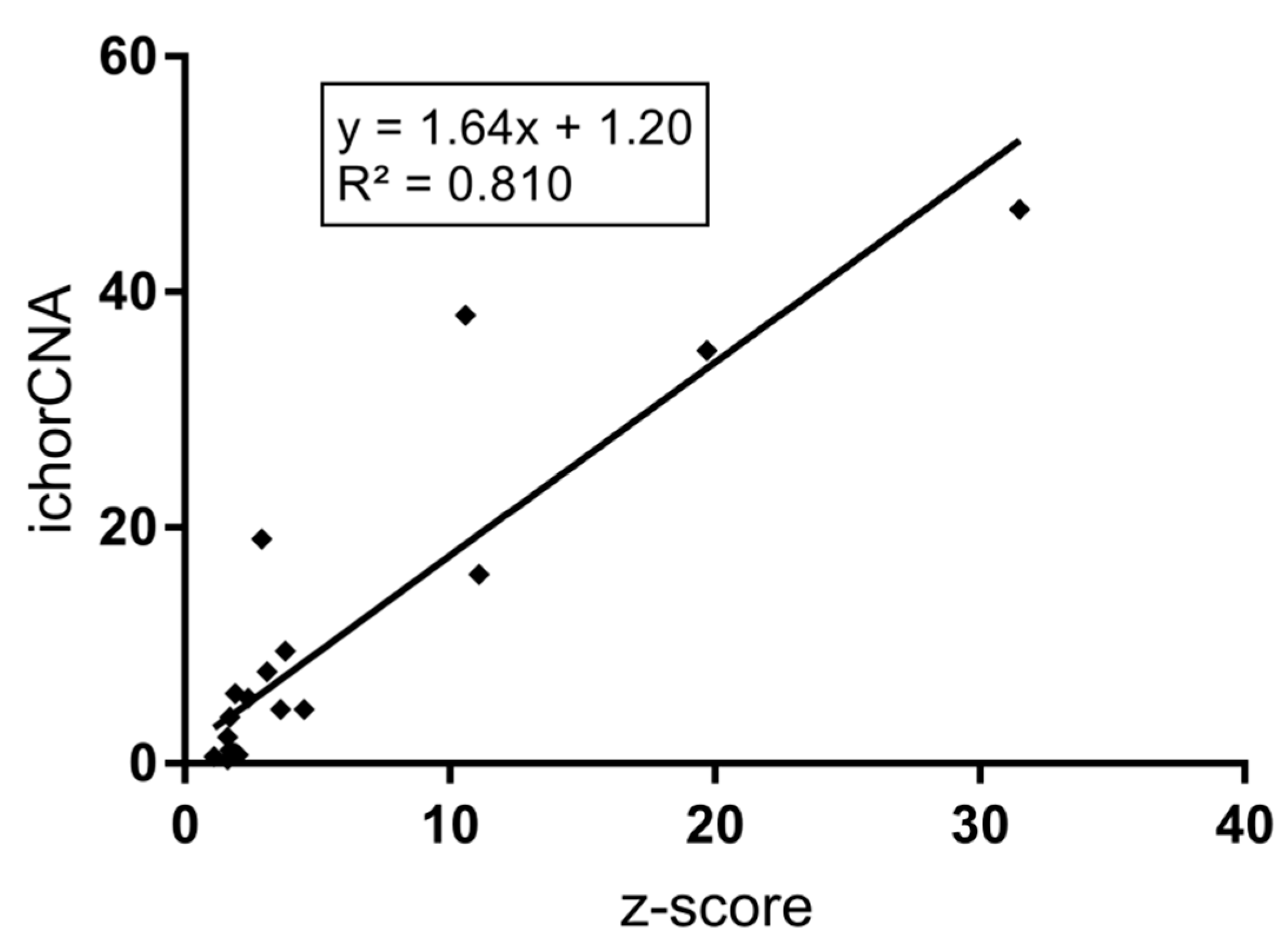
2.2. SCNA Analysis in cfDNA and Evaluation of the Tumor Level
2.3. cfDNA Tumor Fraction Could Predict Survival
3. Discussion
4. Materials and Methods
4.1. Clinicopathologic Data and Sample Collection
4.2. Fluorescence In Situ Hybridization (FISH)
4.3. Genomic Oncoscan Profile
4.4. Cell-Free DNA Extraction and Quantification
4.5. mFAST-SeqS
4.6. Shallow Whole-Genome Sequencing sWGS (PLASMA-SEQ)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perez-Garcia, J.; Muñoz-Couselo, E.; Soberino, J.; Racca, F.; Cortes, J. Targeting FGFR pathway in breast cancer. Breast 2018, 37, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbauomy Elsheikh, S.; Green, A.R.; Lambros, M.B.K.; Turner, N.C.; Grainge, M.J.; Powe, D.; Ellis, I.O.; Reis-Filho, J.S. FGFR1 amplification in breast carcinomas: A chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007, 9, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelsi-Boyer, V.; Orsetti, B.; Cervera, N.; Finetti, P.; Sircoulomb, F.; Rougé, C.; Lasorsa, L.; Letessier, A.; Ginestier, C.; Monville, F.; et al. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol. Cancer Res. 2005, 3, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Liu, X.; Wang, S.; Zhang, Z.; Wu, Z.; Zhang, X.; Li, J. Prognostic value of FGFR gene amplification in patients with different types of cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e105524. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carene, D.; Tran-Dien, A.; Lemonnier, J.; Dalenc, F.; Levy, C.; Pierga, J.-Y.; Jacot, W.; Canon, J.-L.; Richon, C.; Lacroix, L.; et al. Association between FGFR1 copy numbers, MAP3K1 mutations, and survival in axillary node-positive, hormone receptor-positive, and HER2-negative early breast cancer in the PACS04 and METABRIC studies. Breast Cancer Res. Treat. 2020, 179, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Seo, A.N.; Park, S.Y.; Kim, J.Y.; Park, J.Y.; Yu, J.H.; Ahn, J.-H.; Gong, G. Low prognostic implication of fibroblast growth factor family activation in triple-negative breast cancer subsets. Ann. Surg. Oncol. 2014, 21, 1561–1568. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Tsang, J.Y.S.; Ni, Y.-B.; Chan, S.-K.; Chan, K.-F.; Tse, G.M. FGFR1 is an adverse outcome indicator for luminal A breast cancers. Oncotarget 2016, 7, 5063–5073. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Kim, E.; Choi, Y.; Lee, H.; Kim, Y.; Kim, J.; Kang, E.; Kim, S.-W.; Kim, I.; Park, S. FGFR1 is amplified during the progression of in situ to invasive breast carcinoma. Breast Cancer Res. 2012, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Rueda, O.M.; Sammut, S.-J.; Seoane, J.A.; Chin, S.-F.; Caswell-Jin, J.L.; Callari, M.; Batra, R.; Pereira, B.; Bruna, A.; Ali, H.R.; et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 2019, 567, 399–404. [Google Scholar] [CrossRef]
- Drago, J.Z.; Formisano, L.; Juric, D.; Niemierko, A.; Servetto, A.; Wander, S.A.; Spring, L.M.; Vidula, N.; Younger, J.; Peppercorn, J.; et al. FGFR1 amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor-positive (HR+). Breast Cancer. Clin. Cancer Res. 2019, 25, 6443–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017, 32, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Hayashi, N.; Suzuki, K.; Takimoto, M.; Nakamura, S.; Yamauchi, H. Change in HER2 status after neoadjuvant chemotherapy and the prognostic impact in patients with primary breast cancer. J. Surg. Oncol. 2017, 116, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E. Circulating tumor DNA for modern cancer management. Clin. Chem. 2019, 66, 143–145. [Google Scholar] [CrossRef]
- Heitzer, E.; Ulz, P.; Belic, J.; Gutschi, S.; Quehenberger, F.; Fischereder, K.; Benezeder, T.; Auer, M.; Pischler, C.; Mannweiler, S.; et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med. 2013, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Mohan, S.; Heitzer, E.; Ulz, P.; Lafer, I.; Lax, S.; Auer, M.; Pichler, M.; Gerger, A.; Eisner, F.; Hoefler, G.; et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 2014, 10, e1004271. [Google Scholar] [CrossRef]
- Adalsteinsson, V.A.; Ha, G.; Freeman, S.S.; Choudhury, A.D.; Stover, D.G.; Parsons, H.A.; Gydush, G.; Reed, S.C.; Rotem, D.; Rhoades, J.; et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun. 2017, 8, 1324. [Google Scholar] [CrossRef] [Green Version]
- Hovelson, D.H.; Liu, C.-J.; Wang, Y.; Kang, Q.; Henderson, J.; Gursky, A.; Brockman, S.; Ramnath, N.; Krauss, J.C.; Talpaz, M.; et al. Rapid, ultra low coverage copy number profiling of cell-free DNA as a precision oncology screening strategy. Oncotarget 2017, 8, 89848–89866. [Google Scholar] [CrossRef] [Green Version]
- Stover, D.G.; Parsons, H.A.; Ha, G.; Freeman, S.S.; Barry, W.T.; Guo, H.; Choudhury, A.D.; Gydush, G.; Reed, S.C.; Rhoades, J.; et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J. Clin. Oncol. 2018, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schildhaus, H.-U.; Heukamp, L.C.; Merkelbach-Bruse, S.; Riesner, K.; Schmitz, K.; Binot, E.; Paggen, E.; Albus, K.; Schulte, W.; Ko, Y.-D.; et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Mod. Pathol. 2012, 25, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belic, J.; Koch, M.; Ulz, P.; Auer, M.; Gerhalter, T.; Mohan, S.; Fischereder, K.; Petru, E.; Bauernhofer, T.; Geigl, J.B.; et al. Rapid identification of plasma DNA samples with increased ctDNA levels by a modified FAST-SeqS approach. Clin. Chem. 2015, 61, 838–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulz, P.; Belic, J.; Graf, R.; Auer, M.; Lafer, I.; Fischereder, K.; Webersinke, G.; Pummer, K.; Augustin, H.; Pichler, M.; et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat. Commun. 2016, 7, 12008. [Google Scholar] [CrossRef]
- Tan, G.; Chu, C.; Gui, X.; Li, J.; Chen, Q. The prognostic value of circulating cell-free DNA in breast cancer: A meta-analysis. Medicine (Baltimore) 2018, 97, e0197. [Google Scholar] [CrossRef]
- Suppan, C.; Brcic, I.; Tiran, V.; Mueller, H.D.; Posch, F.; Auer, M.; Ercan, E.; Ulz, P.; Cote, R.J.; Datar, R.H.; et al. Untargeted assessment of tumor fractions in plasma for monitoring and prognostication from metastatic breast cancer patients undergoing systemic treatment. Cancers 2019, 11, 1171. [Google Scholar] [CrossRef] [Green Version]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Gevensleben, H.; Garcia-Murillas, I.; Graeser, M.K.; Schiavon, G.; Osin, P.; Parton, M.; Smith, I.E.; Ashworth, A.; Turner, N.C. Noninvasive detection of HER2 amplification with plasma DNA digital PCR. Clin. Cancer Res. 2013, 19, 3276–3284. [Google Scholar] [CrossRef] [Green Version]
- Pearson, A.; Smyth, E.; Babina, I.S.; Herrera-Abreu, M.T.; Tarazona, N.; Peckitt, C.; Kilgour, E.; Smith, N.R.; Geh, C.; Rooney, C.; et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 2016, 6, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Belic, J.; Graf, R.; Bauernhofer, T.; Cherkas, Y.; Ulz, P.; Waldispuehl-Geigl, J.; Perakis, S.; Gormley, M.; Patel, J.; Li, W.; et al. Genomic alterations in plasma DNA from patients with metastasized prostate cancer receiving abiraterone or enzalutamide. Int. J. Cancer 2018, 143, 1236–1248. [Google Scholar] [CrossRef]
- de Wit, S.; Rossi, E.; Weber, S.; Tamminga, M.; Manicone, M.; Swennenhuis, J.F.; Groothuis-Oudshoorn, C.G.M.; Vidotto, R.; Facchinetti, A.; Zeune, L.L.; et al. Single tube liquid biopsy for advanced non-small cell lung cancer. Int. J. Cancer 2019, 144, 3127–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gru, A.A.; Allred, D.C. FGFR1 amplification and the progression of non-invasive to invasive breast cancer. Breast Cancer Res. 2012, 14, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piqueret-Stephan, L.; Marcaillou, C.; Reyes, C.; Honoré, A.; Letexier, M.; Gentien, D.; Droin, N.; Lacroix, L.; Scoazec, J.-Y.; Vielh, P. Massively parallel DNA sequencing from routinely processed cytological smears. Cancer Cytopathol. 2016, 124, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.M.; Oumie, A.; Togneri, F.S.; Vasques, F.R.; Hau, D.; Taylor, M.; Tinkler-Hundal, E.; Southward, K.; Medlow, P.; McGreeghan-Crosby, K.; et al. Cross-laboratory validation of the OncoScan® FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med. Genomics 2015, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Commo, F.; Guinney, J.; Ferté, C.; Bot, B.; Lefebvre, C.; Soria, J.-C.; André, F. rCGH: A comprehensive array-based genomic profile platform for precision medicine. Bioinformatics 2016, 32, 1402–1404. [Google Scholar] [CrossRef] [Green Version]
- Kinde, I.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. FAST-SeqS: A simple and efficient method for the detection of aneuploidy by massively parallel sequencing. PLoS ONE 2012, 7, e41162. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Perakis, S.O.; Ulz, P.; Mohan, S.; Riedl, J.M.; Talakic, E.; Lax, S.; Tötsch, M.; Hoefler, G.; Bauernhofer, T.; et al. Cell-free DNA analysis reveals POLR1D-mediated resistance to bevacizumab in colorectal cancer. Genome Med. 2020, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Lu, L.; Zhou, Z. CNV Detection from Circulating Tumor DNA in Late Stage Non-Small Cell Lung Cancer Patients. Genes 2019, 10, 926. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caption | FGFR1 Amplified | FGFR1 Amplified | Total |
|---|---|---|---|
| N = 20 | N = 80 | N = 100 | |
| AGE (years) median | 56 (28–78) | 60 (32–83) | 60 (28–83) |
| <50 year | 4/20 | 25/80 | 29 |
| DFI (months) median | 35 (0–183) | 31 (0–227) | 33 (0–227) |
| IDC | 18 (90%) | 71 (89%) | 89 |
| Lobular | 2 (10%) | 9 (11%) | 11 |
| Primary tumor grade * | - | - | - |
| 1 | 0 | 8 | 8 |
| 2 | 9 | 32 | 41 |
| 3 | 11 | 36 | 47 |
| ER+/− | 19/1 | 69/10 | 88/11 |
| PR+/− | 13/7 | 47/27 | 60/34 |
| TNBC ** | 1 | 10 (out of 79) | 11 (out of 99) |
| Visceral metastasis y/n | 15/5 | 40/40 | 55/45 |
| Metastatic sites >2 y/n | 10/10 | 23/57 | 33/67 |
| Caption | Evaluation Criteria * FGFR1 was Amplified, If FGFR1/CEN8 Ratio was ≥2.0 and/or Average Number of FGFR1 Signals/Tumor Cell was ≥6.0 | Additional Evaluation Criteria (Low Amplification) FGFR1 was Amplified, If the Percentage of Tumor Cells Containing ≥5 Gene Signals was ≥50% and/or Percentage of Tumor Cells Containing ≥15 Gene Signals or Large Clusters was ≥10% | |||||
|---|---|---|---|---|---|---|---|
| Case No. (Patient ID) | Target Gene/Centromere Ratio | Average Number of Target Gene Signals/Nucleus | Average Number of Centromere Signals/Nucleus | FGFR1 Status | Percentage of Tumor Cells Containing ≥5 Gene Signals (Quotient) | Percentage of Tumor Cells Containing ≥15 Gene Signals or Large Clusters(Quotient) | FGFR1 Status |
| 446894 | 2.3 | 6.3 | 2.8 | amplified | 0.8 | 0 | amplified |
| 525973 | 1.9 | 7.7 | 4.1 | amplified | 0.9 | 0.1 | amplified |
| 529014 | 3.4 | 7.2 | 2.1 | amplified | 0.9 | 0 | amplified |
| 531084 | 2.5 | 6.3 | 2.5 | amplified | 0.8 | 0 | amplified |
| 537264 | 1.1 | 7.5 | 7.1 | amplified | 0.9 | 0.1 | amplified |
| 542827 | 4.5 | 11.6 | 2.6 | amplified | 1.0 | 0.3 | amplified |
| 548602 | 6.7 | 16.2 | 2.4 | amplified | 1.0 | 0.6 | amplified |
| 550066 | 1.9 | 5.9 | 3.1 | balanced | 0.8 | 0 | amplified |
| 557513 | 5.5 | 10.8 | 2.0 | amplified | 1.0 | 0.3 | amplified |
| 562315 | 8.4 | 25.1 | 3.0 | amplified | 1.0 | 0.9 | amplified |
| 587002 | 3.4 | 15.6 | 4.6 | amplified | 1.0 | 0.7 | amplified |
| 594270 | 2.8 | 11.6 | 4.2 | amplified | 1.0 | 0.3 | amplified |
| 595763 | 2.5 | 10.5 | 4.2 | amplified | 1.0 | 0.1 | amplified |
| 599471 | 5.4 | 9.8 | 1.8 | amplified | 1.0 | 0.1 | amplified |
| 600575 | 6.2 | 12.8 | 2.0 | amplified | 1.0 | 0.4 | amplified |
| 615064 | 1.9 | 4.8 | 2.6 | balanced | 0.6 | 0 | amplified |
| 621823 | 5.6 | 12.7 | 2.2 | amplified | 1.0 | 0.4 | amplified |
| 623854 | 4.6 | 7.8 | 1.7 | amplified | 0.7 | 0.1 | amplified |
| 630510 | 4.2 | 5.3 | 1.3 | amplified | 0.6 | 0 | amplified |
| 631774 | 1.9 | 5.1 | 2.7 | balanced | 0.6 | 0 | amplified |
| Case No. (Patient ID) | FGFR1 Status Oncoscan | FGFR1 Copy Number Oncoscan | Concordance with FISH | Biopsies-FISH | Copy Number: Average Number of Target Gene Signals/Nucleus |
|---|---|---|---|---|---|
| 529014 | Gain | 3 | Yes | Amplified | 7.2 |
| 531084 | Gain | 4 | Yes | Amplified | 6.3 |
| 542827 | No gain | 2 | No | Amplified | 11.6 |
| 548602 | Gain | 5 | Yes | Amplified | 16.2 |
| 557513 | Gain | 8 | Yes | Amplified | 10.8 |
| 562315 | Gain | 24 | Yes | Amplified | 25.1 |
| 587002 | Gain | 5 | Yes | Amplified | 15.6 |
| 594270 | Gain | 16 | Yes | Amplified | 11.6 |
| 600575 | Gain | 7 | Yes | Amplified | 12.8 |
| 623854 | Gain | 15 | Yes | Amplified | 7.8 |
| 630510 | Gain | 5 | Yes | Amplified | 5.3 |
| Case No. (Patient ID) | Biopsies-FISH | Oncoscan-Curie | mFAST-SeqS Z-Score | ichorCNA Tumor Fraction (%) | FGFR1 Amplification Calling Based on Plasma-Seq Data |
|---|---|---|---|---|---|
| 529014 | Amplified | Gain | 1.6 | 0.35 | - |
| 529773 | Balanced | No Data | 10.6 | 38 | - |
| 530077 | Balanced | No Data | 1.7 | 1.15 | - |
| 531084 | Amplified | Gain | 4.5 | 4.6 | + |
| 542827 | Amplified | No gain | 1.6 | 1.2 | - |
| 544065 | Balanced | No Data | 31.5 | 47 | - |
| 548696 | Balanced | No Data | 2.9 | 19 | - |
| 557513 | Amplified | Gain | 1.7 | 3.9 | - |
| 562315 | Amplified | Gain | 1.9 | 0.86 | + |
| 568895 | Balanced | No Data | 1.1 | 0.6 | - |
| 578098 | Balanced | No Data | 2 | 0.73 | - |
| 587002 | Amplified | Gain | 3.8 | 9.5 | + |
| 590243 | Balanced | No Data | 1.9 | 5.9 | - |
| 594270 | Amplified | Gain | 2.4 | 5.5 | + |
| 600575 | Amplified | Gain | 3.6 | 4.6 | + |
| 612988 | Balanced | No Data | 1.8 | 0.7 | - |
| 615064 | Amplified low | No Data | 19.7 | 35 | + |
| 615972 | Balanced | No Data | 3.1 | 7.77 | - |
| 623854 | Amplified | Gain | 11.1 | 16 | + |
| 628696 | Balanced | No Data | 1.6 | 2.2 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourrier, C.; Pierga, J.-Y.; Xuereb, L.; Salaun, H.; Proudhon, C.; Speicher, M.R.; Belic, J.; Heitzer, E.; Lockhart, B.P.; Guigal-Stephan, N. Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival. Cancers 2020, 12, 1481. https://doi.org/10.3390/cancers12061481
Bourrier C, Pierga J-Y, Xuereb L, Salaun H, Proudhon C, Speicher MR, Belic J, Heitzer E, Lockhart BP, Guigal-Stephan N. Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival. Cancers. 2020; 12(6):1481. https://doi.org/10.3390/cancers12061481
Chicago/Turabian StyleBourrier, Chantal, Jean-Yves Pierga, Laura Xuereb, Hélène Salaun, Charlotte Proudhon, Michael R. Speicher, Jelena Belic, Ellen Heitzer, Brian Paul Lockhart, and Nolwen Guigal-Stephan. 2020. "Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival" Cancers 12, no. 6: 1481. https://doi.org/10.3390/cancers12061481
APA StyleBourrier, C., Pierga, J.-Y., Xuereb, L., Salaun, H., Proudhon, C., Speicher, M. R., Belic, J., Heitzer, E., Lockhart, B. P., & Guigal-Stephan, N. (2020). Shallow Whole-Genome Sequencing from Plasma Identifies FGFR1 Amplified Breast Cancers and Predicts Overall Survival. Cancers, 12(6), 1481. https://doi.org/10.3390/cancers12061481