Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique

 , , ,
, , ,
Abstract
1. Introduction
2. Results
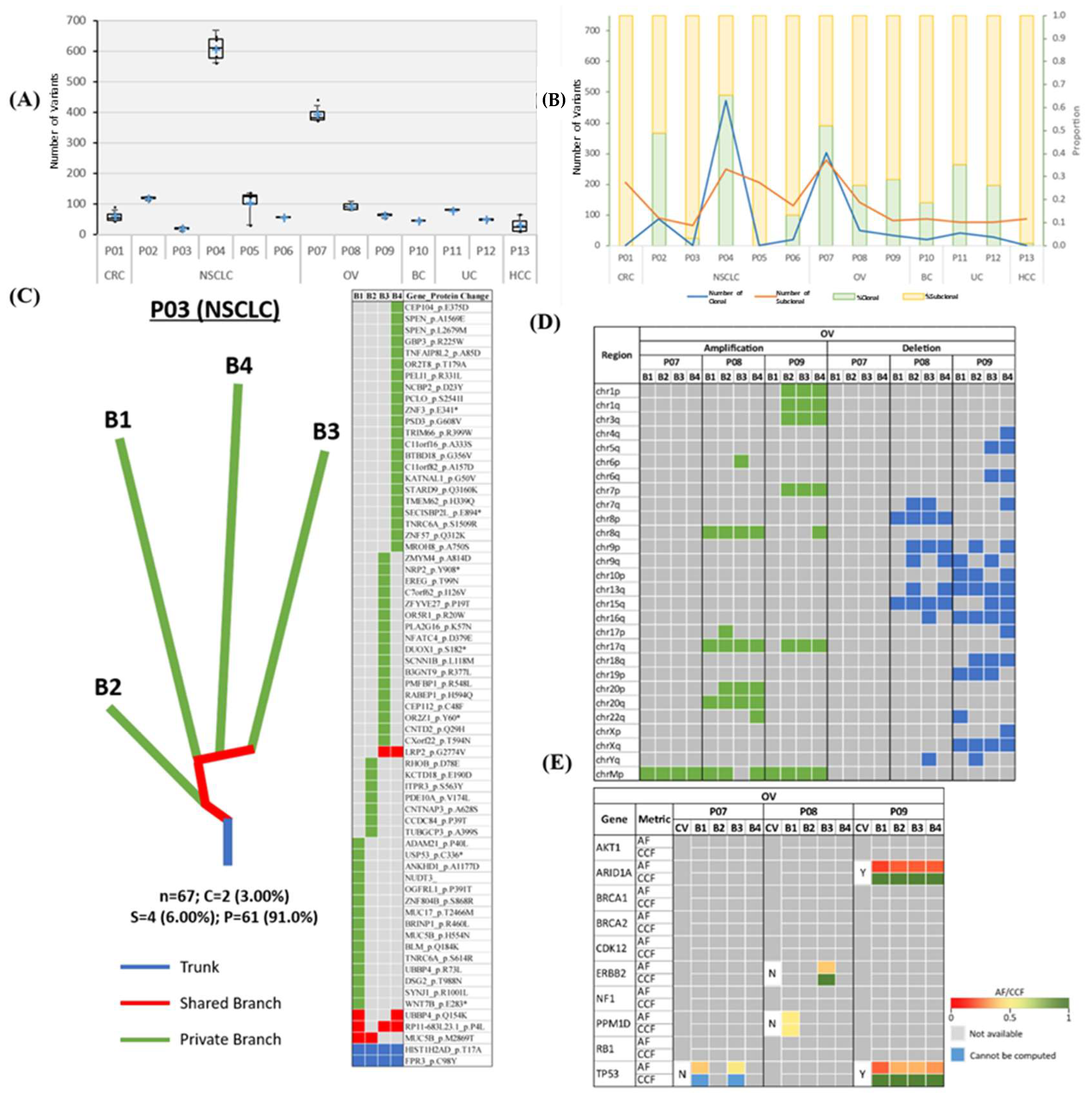
2.1. Tumour Variant Load
2.2. Intratumor Heterogeneity
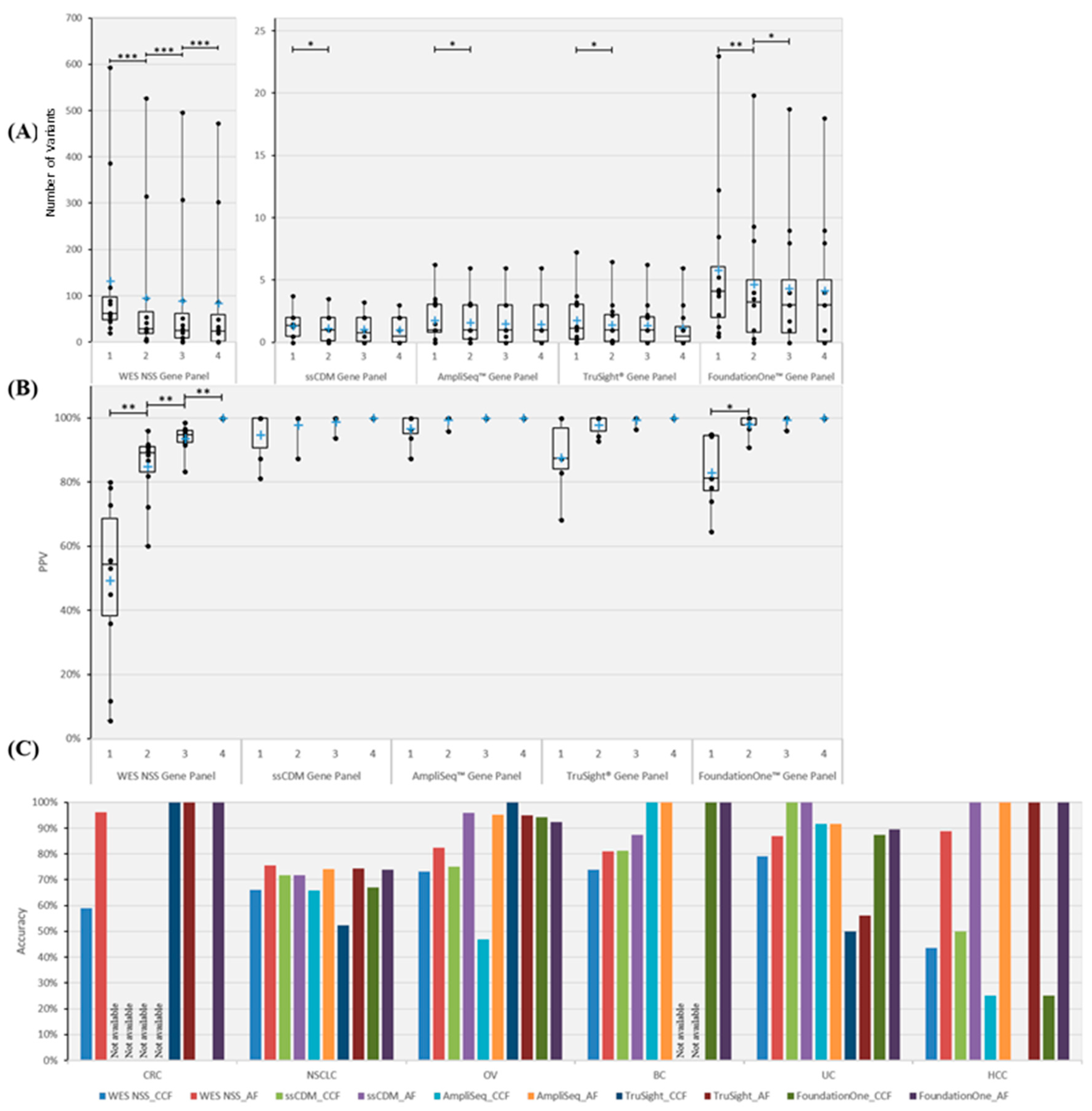
2.3. Statistical Saturation Analysis
2.4. Prediction of Truncal Mutations
2.5. Clinical Therapeutic Intervention
3. Discussion
4. Materials and Methods
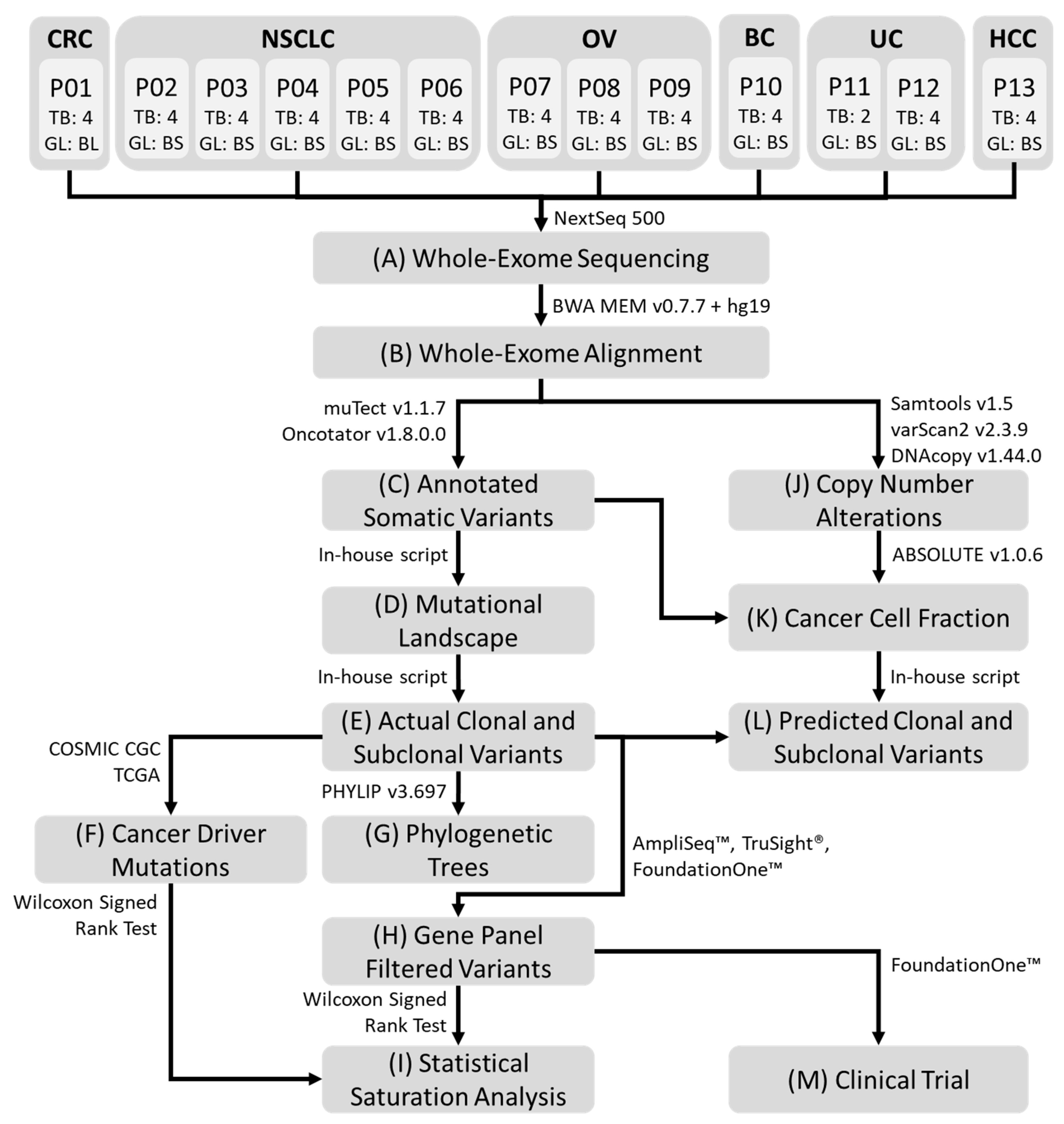
4.1. Patients and Specimens Collection
4.2. Whole-Exome Sequencing
4.3. Sequencing Reads Alignment and Somatic Variant Detection
4.4. Copy Number Alterations
4.5. Cancer Gene Panels
4.6. Construction of Phylogenetic Trees
4.7. Statistical Saturation Analysis
4.8. Cancer Cell Fraction and Allele Frequency
4.9. Prediction of Truncal Mutations
4.10. Assessing Targeted Therapy Outcomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Sample | Patient | Patient label | Disease | ABSOLUTE_call Status (Called = Clonal) | ABSOLUTE_purity | ABSOLUTE_ploidy | ABSOLUTECancer DNA Faction | ABSOLUTECoverage for 80% Power | VAF Range-All Variants | VAF Range-Putative Truncal Variants | VAF Range-Private Variants | VAF Range-Branch Variants | Mclust.wCN.Cluster# | Mclust.noCN.Cluster# | VAF_ROC_AUC_TruncalVsNonTruncal | VAF_YoudenThreshold_TruncalVsNonTruncal | VAF_YoudenThreshold_TruncalVsNonTruncal_Accuracy | Mclust.cn.threshold_clusterWHighestVAF | Mclust.cn.threshold_TruncalVsNonTruncal_Accuracy | Remark |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RE3F1 | RE3 | P01 | CRC | called | 0.25 | 5.86 | 0.49 | 64 | 0.14038 | NaN | 0.135531915 | 0.2446 | 2 | 2 | NaN | NaN | NaN | 0.241285714 | NaN | No Truncal Mutation detected |
| RE3F2 | RE3 | P01 | CRC | called | 0.21 | 1.94 | 0.2 | 52 | 0.134576087 | NaN | 0.099253968 | 0.152966942 | 1 | 2 | NaN | NaN | NaN | 0.1086 | NaN | No Truncal Mutation detected |
| RE3F3 | RE3 | P01 | CRC | called | 0.28 | 4.02 | 0.44 | 49 | 0.146770833 | NaN | 0.143824176 | 0.2004 | 2 | 2 | NaN | NaN | NaN | 0.348666667 | NaN | No Truncal Mutation detected |
| RE3F4 | RE3 | P01 | CRC | high non-clonal | 1 | 2.06 | 1 | 8 | 0.383224409 | NaN | 0.30389313 | 0.467715447 | 2 | 2 | NaN | NaN | NaN | 0.447333333 | NaN | No Truncal Mutation detected |
| RE4F1 | RE4 | P02 | LUNG | called | 0.39 | 3.83 | 0.55 | 36 | 0.183591928 | 0.191490909 | 0.163944444 | 0.1565 | 2 | 2 | 0.675130617 | 0.076 | 62.27795193 | 0.419666667 | 50.49111808 | --- |
| RE4F2 | RE4 | P02 | LUNG | called | 0.5 | 3.81 | 0.65 | 29 | 0.205012195 | 0.226848485 | 0.145166667 | 0.177076923 | 2 | 2 | 0.688477366 | 0.091 | 68.24915825 | 0.2135 | 57.35129068 | --- |
| RE4F3 | RE4 | P02 | LUNG | called | 0.22 | 5.43 | 0.43 | 68 | 0.176941909 | 0.192266667 | 0.123045455 | 0.17203125 | 1 | 3 | 0.680582137 | 0.089 | 68.12599681 | 0.20575 | 55.2830941 | --- |
| RE4F4 | RE4 | P02 | LUNG | called | 0.42 | 3.85 | 0.58 | 34 | 0.180034043 | 0.194939394 | 0.1438125 | 0.145815789 | 2 | 2 | 0.714545455 | 0.096 | 67.70562771 | 0.5995 | 49.89177489 | --- |
| RE6F1 | RE6 | P03 | LUNG | called | 0.2 | 4.03 | 0.33 | 66 | 0.106431818 | 0.156 | 0.085482759 | 0.1505 | 0 | 2 | 0.659375 | 0.364 | 50 | 0.173 | 68.75 | Only 2 Truncal Mutations detected |
| RE6F2 | RE6 | P03 | LUNG | called | 0.16 | 1.96 | 0.16 | 66 | 0.106794118 | 0.12125 | 0.102037037 | 0.130333333 | 0 | 3 | 0.575 | 0.5 | 50 | 0.1345 | 63.33333333 | Only 2 Truncal Mutations detected |
| RE6F3 | RE6 | P03 | LUNG | called | 0.23 | 2.12 | 0.24 | 47 | 0.105585366 | 0.236 | 0.0839 | 0.124 | 0 | 2 | 0.692567568 | 0.524 | 62.5 | 0.327333333 | 73.64864865 | Only 2 Truncal Mutations detected |
| RE6F4 | RE6 | P03 | LUNG | called | 0.16 | 1.07 | 0.09 | 61 | 0.106481481 | 0.21825 | 0.079666667 | 0.191375 | 1 | 2 | 0.7525 | 0.52 | 50 | 0.072 | 60.5 | Only 2 Truncal Mutations detected |
| RE8F1 | RE8 | P04 | LUNG | called | 0.32 | 4.21 | 0.49 | 46 | 0.222009821 | 0.225307027 | 0.210836364 | 0.204115108 | 2 | 3 | 0.589563247 | 0.052 | 50.40747256 | 0.296918919 | 53.80586048 | --- |
| RE8F2 | RE8 | P04 | LUNG | high non-clonal | 0.58 | 4.34 | 0.75 | 28 | 0.29151633 | 0.316179459 | 0.201833333 | 0.234288194 | 2 | 3 | 0.655537557 | 0.084 | 55.15448764 | 0.565733333 | 54.26535877 | --- |
| RE8F3 | RE8 | P04 | LUNG | high entropy | 0.46 | 4.39 | 0.65 | 35 | 0.255151779 | 0.275265946 | 0.156716418 | 0.210709559 | 2 | 3 | 0.658003466 | 0.083 | 53.60034914 | 0.359983051 | 57.10528998 | --- |
| RE8F4 | RE8 | P04 | LUNG | called | 0.27 | 3.33 | 0.38 | 47 | 0.192184165 | 0.198313514 | 0.113677419 | 0.187166667 | 3 | 4 | 0.619140554 | 0.049 | 50.58159754 | 0.332363636 | 51.06664348 | --- |
| RE9F1 | RE9 | P05 | LUNG | called | 0.22 | 3.87 | 0.36 | 58 | 0.152 | 0.408 | 0.117938776 | 0.157363095 | 1 | 3 | NaN | NaN | NaN | 0.117214286 | NaN | No Truncal Mutation detected |
| RE9F2 | RE9 | P05 | LUNG | called | 0.31 | 5.63 | 0.56 | 55 | 0.229921642 | 0.414333333 | 0.141388235 | 0.268655556 | 2 | 3 | NaN | NaN | NaN | 0.416 | NaN | No Truncal Mutation detected |
| RE9F3 | RE9 | P05 | LUNG | called | 0.25 | 3.94 | 0.4 | 53 | 0.12454386 | 0.407 | 0.111265306 | 0.0852 | 2 | 2 | NaN | NaN | NaN | 0.08 | NaN | No Truncal Mutation detected |
| RE9F4 | RE9 | P05 | LUNG | called | 0.3 | 4.85 | 0.51 | 51 | 0.156137339 | 0.408333333 | 0.112022222 | 0.162778378 | 1 | 2 | NaN | NaN | NaN | 0.1255 | NaN | No Truncal Mutation detected |
| RE10F1 | RE10 | P06 | LUNG | called | 0.35 | 6.42 | 0.63 | 55 | 0.233589474 | 0.420242424 | 0.106076923 | 0.2807 | 2 | 4 | 0.9335 | 0.2235 | 89.71 | 0.4315 | 71.11 | --- |
| RE10F2 | RE10 | P06 | LUNG | called | 0.24 | 3.98 | 0.38 | 56 | 0.154539216 | 0.229878788 | 0.117761905 | 0.126333333 | 2 | 2 | 0.8304787 | 0.167 | 80.57 | 0.193142857 | 72.99 | --- |
| RE10F3 | RE10 | P06 | LUNG | called | 0.26 | 5.3 | 0.48 | 60 | 0.203761905 | 0.402121212 | 0.103064516 | 0.1735 | 2 | 2 | 0.962121212 | 0.311 | 88.13131313 | 0.336714286 | 82.07070707 | --- |
| RE10F4 | RE10 | P06 | LUNG | called | 0.35 | 4.34 | 0.54 | 43 | 0.203847826 | 0.376272727 | 0.08216 | 0.247666667 | 2 | 2 | 0.942218798 | 0.273 | 91.88495121 | 0.332857143 | 76.91319979 | --- |
| RE5F1 | RE5 | P07 | OV | non-aneuploid | NaN | NaN | NaN | NaN | 0.341515366 | 0.456792381 | 0.119782946 | 0.288920635 | 4 | 3 | 0.953009939 | 0.278 | 93.77748109 | 0.547 | 55.3600356 | --- |
| RE5F2 | RE5 | P07 | OV | non-aneuploid | NaN | NaN | NaN | NaN | 0.351859281 | 0.385497143 | 0.176089109 | 0.354071429 | 2 | 3 | 0.767505828 | 0.188 | 76.35164835 | 0.364184211 | 67.12687313 | --- |
| RE5F3 | RE5 | P07 | OV | non-aneuploid | NaN | NaN | NaN | NaN | 0.372459689 | 0.42759619 | 0.138438095 | 0.315649351 | 1 | 3 | 0.858644689 | 0.258 | 82.34065934 | 0.378807692 | 79.35531136 | --- |
| RE5F4 | RE5 | P07 | OV | non-aneuploid | NaN | NaN | NaN | NaN | 0.391212257 | 0.431544762 | 0.17428125 | 0.3839375 | 2 | 6 | 0.81843254 | 0.28 | 78.18055556 | 0.424096774 | 68.52380952 | --- |
| RE11F1 | RE11 | P08 | OV | called | 0.27 | 3.6 | 0.4 | 48 | 0.182677632 | 0.214707071 | 0.109380952 | 0.174272727 | 2 | 3 | 0.825995807 | 0.093 | 80.05526968 | 0.458 | 53.09700781 | --- |
| RE11F2 | RE11 | P08 | OV | called | 0.25 | 3.02 | 0.34 | 48 | 0.22096988 | 0.273212121 | 0.119104167 | 0.206105263 | 4 | 3 | 0.80830695 | 0.122 | 79.05924921 | 0.383 | 57.59837178 | --- |
| RE11F3 | RE11 | P08 | OV | called | 0.25 | 5.06 | 0.46 | 59 | 0.219164894 | 0.279626263 | 0.135373134 | 0.202272727 | 1 | 3 | 0.806037907 | 0.123 | 79.04891613 | 0.15175 | 77.1422086 | --- |
| RE11F4 | RE11 | P08 | OV | called | 0.22 | 4.74 | 0.4 | 65 | 0.225337079 | 0.294050505 | 0.1038 | 0.220416667 | 1 | 3 | 0.837424882 | 0.152 | 80.39253292 | 0.2655 | 67.40825981 | --- |
| RE13F1 | RE13 | P09 | OV | called | 0.47 | 5.47 | 0.71 | 41 | 0.196666667 | 0.242578125 | 0.126136364 | 0.215 | 2 | 2 | 0.783166274 | 0.138 | 78.19870283 | 0.370333333 | 55.76356132 | --- |
| RE13F2 | RE13 | P09 | OV | called | 0.32 | 5.19 | 0.55 | 50 | 0.254631944 | 0.317359375 | 0.103151515 | 0.275574468 | 1 | 3 | 0.741113281 | 0.229 | 71.875 | 0.259411765 | 66.40625 | --- |
| RE13F3 | RE13 | P09 | OV | called | 0.31 | 5.3 | 0.55 | 52 | 0.267333333 | 0.34478125 | 0.112880952 | 0.299893617 | 1 | 3 | 0.738851826 | 0.172 | 72.05933989 | 0.222090909 | 64.9315309 | --- |
| RE13F4 | RE13 | P09 | OV | called | 0.33 | 5.21 | 0.57 | 49 | 0.295875969 | 0.355515625 | 0.172761905 | 0.267886364 | 1 | 2 | 0.705288462 | 0.145 | 69.19471154 | 0.229583333 | 64.42307692 | --- |
| RE7F1 | RE7 | P10 | BRCA | called | 0.82 | 2.3 | 0.84 | 11 | 0.31790099 | 0.44826087 | 0.1773125 | 0.425285714 | 1 | 2 | 0.832806324 | 0.289 | 82.74703557 | 0.325 | 79.48616601 | --- |
| RE7F2 | RE7 | P10 | BRCA | called | 0.78 | 2.32 | 0.81 | 12 | 0.3082 | 0.423347826 | 0.131810811 | 0.483857143 | 1 | 2 | 0.863142292 | 0.304 | 86.56126482 | 0.2225 | 83.05335968 | --- |
| RE7F3 | RE7 | P10 | BRCA | called | 0.29 | 5.98 | 0.55 | 59 | 0.311932584 | 0.433630435 | 0.11159375 | 0.385818182 | 1 | 2 | 0.861223458 | 0.3 | 89.76238625 | 0.269 | 86.27401416 | --- |
| RE7F4 | RE7 | P10 | BRCA | called | 0.22 | 3.61 | 0.33 | 59 | 0.19171134 | 0.237978261 | 0.143681818 | 0.189571429 | 1 | 2 | 0.805626598 | 0.132 | 83.01364024 | 0.113666667 | 81.15942029 | --- |
| RE12F1 | RE12 | P11 | UCEC | high non-clonal | 0.63 | 2.24 | 0.66 | 14 | 0.221503448 | * 0.323226666666667 | 0.112514286 | NaN | 2 | 2 | 0.903142857 | 0.164 | 88.28571429 | 0.31075 | 73.85714286 | --- |
| RE12F4 | RE12 | P11 | UCEC | called | 0.66 | 4.45 | 0.81 | 26 | 0.267685185 | * 0.4072 | 0.14737931 | NaN | 3 | 3 | 0.863984674 | 0.194 | 82.59770115 | 0.9165 | 52.09195402 | --- |
| RE14F1 | RE14 | P12 | UCEC | called | 0.2 | 4.62 | 0.37 | 68 | 0.217402299 | 0.27652 | 0.110096774 | 0.279166667 | 4 | 3 | 0.885135135 | 0.121 | 86.83783784 | 0.518 | 52.2972973 | --- |
| RE14F2 | RE14 | P12 | UCEC | called | 0.26 | 5.79 | 0.51 | 62 | 0.254321839 | 0.30418 | 0.1312 | 0.303083333 | 2 | 3 | 0.817567568 | 0.178 | 82.83783784 | 0.5225 | 51.94594595 | --- |
| RE14F3 | RE14 | P12 | UCEC | called | 0.23 | 4.9 | 0.43 | 62 | 0.295 | 0.4168 | 0.101848485 | 0.3215 | 1 | 3 | 0.882173913 | 0.174 | 85.04347826 | 0.4595 | 60.65217391 | --- |
| RE14F4 | RE14 | P12 | UCEC | called | 0.22 | 5.48 | 0.44 | 68 | 0.295521277 | 0.423 | 0.110857143 | 0.305444444 | 1 | 3 | 0.888636364 | 0.197 | 88.90909091 | 0.495 | 63.72727273 | --- |
| RE15F1 | RE15 | P13 | LIHC | called | 0.43 | 4.05 | 0.61 | 35 | 0.266176 | 0.35 | 0.225596774 | 0.304672131 | 1 | 3 | 0.62601626 | 0.75 | 50 | 0.2527 | 46.54471545 | Only 1 Truncal Mutation detected |
| RE15F2 | RE15 | P13 | LIHC | called | 0.23 | 1.96 | 0.23 | 46 | 0.179296296 | 0.2475 | 0.176842105 | 0.164333333 | 0 | 4 | 0.72 | 0.667 | 50 | 0.428666667 | 73 | Only 1 Truncal Mutation detected |
| RE15F3 | RE15 | P13 | LIHC | called | 0.23 | 2.04 | 0.23 | 47 | 0.166075949 | 0.3635 | 0.15 | 0.163171875 | 0 | 0 | 0.948051948 | 1 | 50 | NaN | NaN | Only 1 Truncal Mutation detected |
| RE15F4 | RE15 | P13 | LIHC | called | 0.2 | 3.91 | 0.32 | 66 | 0.1189 | 0.28 | 0.084888889 | 0.1479 | 0 | 0 | 0.946428571 | 0.667 | 50 | NaN | NaN | Only 1 Truncal Mutation detected |
References
- Jamal-Hanjani, M.; Wilson, G.A. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Evolutionary determinants of cancer. Cancer Discov. 2015, 5, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012, 13, 178–185. [Google Scholar] [CrossRef]
- Morris, L.G.T.; Riaz, N. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016, 7, 10051–10063. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef]
- Gerlinger, M.; Horswell, S. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Yap, T.A.; Gerlinger, M. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127–137. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Loeb, L.A. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Au, J.S.-K. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Heong, V.; Syn, N.L. Value of a molecular screening program to support clinical trial enrollment in Asian cancer patients: The Integrated Molecular Analysis of Cancer (IMAC) Study. Int. J. Cancer 2018, 142, 1890–1900. [Google Scholar] [CrossRef]
- Wang, S.; Cang, S. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73, 276–284. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- Jovelet, C.; Ileana, E. Circulating Cell-Free Tumor DNA Analysis of 50 Genes by Next-Generation Sequencing in the Prospective MOSCATO Trial. Clin. Cancer Res. 2016, 22, 2960–2968. [Google Scholar] [CrossRef]
- Marchetti, A.; Palma, J.F. Early Prediction of Response to Tyrosine Kinase Inhibitors by Quantification of EGFR Mutations in Plasma of NSCLC Patients. J. Thorac. Oncol. 2015, 10, 1437–1443. [Google Scholar] [CrossRef]
- Sorensen, B.S.; Wu, L. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer 2014, 120, 3896–3901.20. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.H.; Lichtenstein, L. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar]
- Seshan, V.E.; Olshen, A. DNAcopy: DNA copy number data analysis. R package version 1.62.0. Available online: https://bioconductor.org/biocLite.R (accessed on 14 August 2017).
- Futreal, P.A.; Coin, L. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Wellcome Sanger Institute. Cancer Gene Census. 2011. Available online: http://www.sanger.ac.uk/genetics/CGP/Census/ (accessed on 10 June 2018).
- Lawrence, M.S.; Stojanov, P. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Thermo Fisher. Ion AmpliSeq Cancer Hotspot Panel v2. Available online: https://www.thermofisher.com/order/catalog/product/4475346 (accessed on 28 November 2018).
- Illumina. TruSight® Cancer Panel. Available online: https://www.illumina.com/products/by-type/clinical-research-products/trusight-cancer.html (accessed on 26 July 2018).
- Foundation Medicine. FoundationOneTM Cancer Gene Panel. Available online: https://www.foundationmedicine.com/genomic-testing/foundation-one (accessed on 11 August 2018).
- Felsenstein, J. PHYLIP (Phylogeny Inference Package) Version 3.6; Distributed by the Author; Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2005; Available online: http://evolution.genetics.washington.edu/phylip.html (accessed on 15 August 2017).
- Von Hoff, D.D.; Stephenson, J.J. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J. Clin. Oncol. 2010, 28, 4877–4883. [Google Scholar] [CrossRef]



| Cancer Type | Patient | Age | Sex | No. of MRTB Samples with Abnormality of Interest | No. of MRTB Samples that CCF Metric Classified as Clonal | Targeted Abnormality | Therapeutic Intervention | PFS (Months) | PFS Ratio | Radiological RECIST (v1.1) Response | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Initial Therapy | Actionable Truncal Mutation-Directed Therapy | ||||||||||
| NSCLC | P06 | 74 | M | 4/4 | 3/4 | EGFR T790M | T790M inhibitor | 2.5 | 25.5 | 10.2 | PR |
| NSCLC | P05 | 43 | M | 3/4 | NA | EGFR T790M | T790M inhibitor | 2.1 | 3.6 | 1.71 | SD |
| BC | P10 | 41 | F | 4/4 | 4/4 | PIK3CA H1047R | PI3Kα/β inhibitor | 2 | 1.9 | 0.95 | PD |
| UC | P11 | 46 | F | 2/2 | 2/2 | AKT1 E17K | pan-AKT inhibitor | 4 | 6.1 | 1.53 | SD |
| Panel | CCF | AF |
|---|---|---|
| FoundationOne | 0.92 | 0.13 |
| AmpliSeq | 0.92 | 0.15 |
| TruSight | 0.96 | 0.13 |
| WES | 1 | 0.16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heong, V.; Tay, D.; Goh, S.E.; Wee, B.; Tan, T.Z.; Soo, R.; Pang, B.; Lim, D.; Gopinathan, A.; Ow, S.; et al. Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique. Cancers 2020, 12, 1599. https://doi.org/10.3390/cancers12061599
Heong V, Tay D, Goh SE, Wee B, Tan TZ, Soo R, Pang B, Lim D, Gopinathan A, Ow S, et al. Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique. Cancers. 2020; 12(6):1599. https://doi.org/10.3390/cancers12061599
Chicago/Turabian StyleHeong, Valerie, Darwin Tay, Shane Ee Goh, Bernard Wee, Tuan Zea Tan, Ross Soo, Brendan Pang, Diana Lim, Anil Gopinathan, Samuel Ow, and et al. 2020. "Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique" Cancers 12, no. 6: 1599. https://doi.org/10.3390/cancers12061599
APA StyleHeong, V., Tay, D., Goh, S. E., Wee, B., Tan, T. Z., Soo, R., Pang, B., Lim, D., Gopinathan, A., Ow, S., Chee, C. E., Goh, B. C., Lee, S. C., Yong, W. P., Wong, A., Omar, M. F. M., Soong, R., & Tan, D. S. (2020). Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique. Cancers, 12(6), 1599. https://doi.org/10.3390/cancers12061599