The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer




Abstract
:1. Introduction
2. The Molecular Landscape of Endometrial Cancer
3. Eukaryotic Initiation Factors in Carcinogenesis
4. Clinical Trials on eIFs in Cancer
5. Epithelial Mesenchymal Transition in Endometrial Cancers
6. Eukaryotic Initiation Factors in EMT
7. Clinical Implications of EMT in EC
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sahoo, S.S.; Zhang, X.D.; Hondermarck, H.; Tanwar, P.S. The emerging role of the microenvironment in endometrial cancer. Cancers 2018, 10, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.-B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II Endometrial Cancers: Have They Different Risk Factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Dønnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, K.; Östman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, H.; Li, M.; Xue, J.; Fu, Y.; Yan, L.; Zhao, X. Normal endometrial stromal cells regulate survival and apoptosis signaling through PI3K/AKt/Survivin pathway in endometrial adenocarcinoma cells in vitro. Gynecol. Oncol. 2011, 123, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.T.; Lessey, B.A.; Seppälä, M.; Kaufman, D.G. Effect of normal endometrial stroma on growth and differentiation in Ishikawa endometrial adenocarcinoma cells. Cancer Res. 2002, 62, 79–88. [Google Scholar] [PubMed]
- Sriram, A.; Bohlen, J.; Teleman, A.A. Translation acrobatics: How cancer cells exploit alternate modes of translational initiation. EMBO Rep. 2018, 19, e45947. [Google Scholar] [CrossRef]
- Urick, M.E.; Bell, D.W. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 2019, 19, 510–521. [Google Scholar] [CrossRef]
- Chiu, H.-C.; Li, C.-J.; Yiang, G.-T.; Tsai, A.; Wu, M.-Y. Epithelial to Mesenchymal Transition and Cell Biology of Molecular Regulation in Endometrial Carcinogenesis. J. Clin. Med. 2019, 8, 439. [Google Scholar] [CrossRef] [Green Version]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat. Cell Biol. 2009, 11, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.; Cargnello, M.; Topisirovic, I.; Pelletier, J. Translation Initiation Factors: Reprogramming Protein Synthesis in Cancer. Trends Cell Biol. 2016, 26, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Spilka, R.; Ernst, C.; Mehta, A.K.; Haybaeck, J. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013, 340, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Andreou, A.Z.; Klostermeier, D. The DEAD-box helicase eIF4A. RNA Biol. 2013, 10, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Villa, N.; Do, A.; Hershey, J.W.B.; Fraser, C.S. Human Eukaryotic Initiation Factor 4G (eIF4G) Protein Binds to eIF3c, -d, and -e to Promote mRNA Recruitment to the Ribosome. J. Biol. Chem. 2013, 288, 32932–32940. [Google Scholar] [CrossRef] [Green Version]
- Hinnebusch, A.G.; Lorsch, J.R. The Mechanism of Eukaryotic Translation Initiation: New Insights and Challenges. Cold Spring Harb. Perspect. Biol. 2012, 4, a011544. [Google Scholar] [CrossRef] [Green Version]
- Ruggero, D. Translational Control in Cancer Etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhang, J.-T. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit. Rev. Oncol. Hematol. 2006, 59, 169–180. [Google Scholar] [CrossRef]
- Lazaris-Karatzas, A.; Montine, K.S.; Sonenberg, N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5’ cap. Nature 1990, 345, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Czapiewski, P.; Lapińska-Szumczyk, S.; Majewska, H.; Supernat, A.; Zaczek, A.; Biernat, W.; Golob-Schwarzl, N.; Haybaeck, J. The Prognostic Significance of Eukaryotic Translation Initiation Factors (eIFs) in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; Lee, J.-S.; Kim, S.R.; Lee, Y.-Y.; Kim, C.-J.; Lee, J.-W.; Kim, T.-J.; Lee, J.-H.; Kim, B.-G.; Bae, D.-S. Direct inhibition of eIF4E reduced cell growth in endometrial adenocarcinoma. J. Cancer Res. Clin. Oncol. 2011, 137, 463–469. [Google Scholar] [CrossRef]
- Shi, Z.M.; Liu, Y.N.; Fu, B.; Shen, Y.F.; Li, L.M. Expression profile of eukaryotic translation initiation factor and matrix metalloproteinase 9 in endometrial cancer tissue. J. Biol. Regul. Homeost. Agents 2017, 31, 1053–1059. [Google Scholar] [PubMed]
- Zhang, H.; Li, R.; Li, Y.; Yu, X.; Sun, Q.; Li, A.; Kong, Y. eIF4E-related miR-320a and miR-340-5p inhibit endometrial carcinoma cell metastatic capability by preventing TGF-β1-induced epithelial-mesenchymal transition. Oncol. Rep. 2020, 43, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Mannelqvist, M.; Stefansson, I.M.; Bredholt, G.; Hellem Bø, T.; Øyan, A.M.; Jonassen, I.; Kalland, K.-H.; Salvesen, H.B.; Akslen, L.A. Gene Expression Patterns Related to Vascular Invasion and Aggressive Features in Endometrial Cancer. Am. J. Pathol. 2011, 178, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- EASL. Clinical Practice Guidelines: Management of hepatitis C virus infection. J. Hepatol. 2014, 60, 392–420. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Oh, Y.; Wheler, J.; Naing, A.; Brail, L.; Callies, S.; Andre, V.; Kadam, S.K.; Nasir, A.; et al. A Phase 1 Dose Escalation, Pharmacokinetic, and Pharmacodynamic Evaluation of eIF-4E Antisense Oligonucleotide LY2275796 in Patients with Advanced Cancer. Clin. Cancer Res. 2011, 17, 6582–6591. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.T.; Thompson, P.A.; Nilewski, C.; Sprengeler, P.A.; Sperry, S.; Packard, G.; Michels, T.; Xiang, A.; Tran, C.; Wegerski, C.J.; et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J. Med. Chem. 2020, 63, 5879–5955. [Google Scholar] [PubMed]
- Duffy, A.G.; Makarova-Rusher, O.V.; Ulahannan, S.V.; Rahma, O.E.; Fioravanti, S.; Walker, M.; Abdullah, S.; Raffeld, M.; Anderson, V.; Abi-Jaoudeh, N.; et al. Modulation of tumor eIF4E by antisense inhibition: A phase I/II translational clinical trial of ISIS 183750-an antisense oligonucleotide against eIF4E-in combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int. J. Cancer 2016, 139, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.; Culjkovic-Kraljacic, B.; Bergeron, J.; Caplan, S.; Cocolakis, E.; Lambert, C.; Lau, C.J.; Zahreddine, H.A.; Miller, W.H.; Borden, K.L.B. A phase I trial of ribavirin and low-dose cytarabine for the treatment of relapsed and refractory acute myeloid leukemia with elevated eIF4E. Haematologica 2015, 100, e7–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Tanabe, S.; Aoyagi, K.; Yokozaki, H.; Sasaki, H. Gene expression signatures for identifying diffuse-type gastric cancer associated with epithelial-mesenchymal transition. Int. J. Oncol. 2014, 44, 1955–1970. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.L.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ye, F.; Jiang, X.; Guo, H.; Xie, W.; Zhang, Y.; Sheng, X. Drp1 mediates high glucose-induced mitochondrial dysfunction and epithelial-mesenchymal transition in endometrial cancer cells. Exp. Cell Res. 2020, 389, 111880. [Google Scholar] [CrossRef]
- Xiao, Y.-Y.; Lin, L.; Li, Y.-H.; Jiang, H.-P.; Zhu, L.-T.; Deng, Y.-R.; Lin, D.; Chen, W.; Zeng, C.-Y.; Wang, L.-J.; et al. ZEB1 promotes invasion and metastasis of endometrial cancer by interacting with HDGF and inducing its transcription. Am. J. Cancer Res. 2019, 9, 2314–2330. [Google Scholar]
- Liu, C.; Wang, L.; Jiang, Q.; Zhang, J.; Zhu, L.; Lin, L.; Jiang, H.; Lin, D.; Xiao, Y.; Fang, W.; et al. Hepatoma-derived growth factor and DDX5 promote carcinogenesis and progression of endometrial cancer by activating β-catenin. Front. Oncol. 2019, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.J.; Liang, S.M.; He, P.J.; Zhao, X.B.; Li, M.J.; Geng, F. Dusp6 inhibits epithelial-mesenchymal transition in endometrial adenocarcinoma via ERK signaling pathway. Radiol. Oncol. 2019, 53, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Guo, Q.; Wang, C.; Yan, L.; Fu, Y.; Fan, M.; Zhao, X.; Li, M. Dual-specificity phosphatase 6 (Dusp6), a negative regulator of FGF2/ERK1/2 signaling, enhances 17β-estrodial-induced cell growth in endometrial adenocarcinoma cell. Mol. Cell. Endocrinol. 2013, 376, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, R.; Chi, S.; Zhang, W.; Xiao, C.; Zhou, X.; Zhao, Y.; Wang, H. UBE2C is upregulated by estrogen and promotes epithelial-mesenchymal transition via p53 in endometrial cancer. Mol. Cancer Res. 2020, 18, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, I.C.; Sahoo, S.S.; Kumar, A.; Zhang, H.; Westcott, J.; Aguilar, M.; Cortez, J.D.; Sullivan, S.A.; Xing, C.; Neil Hayes, D.; et al. Fbxw7 is a driver of uterine carcinosarcoma by promoting epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2019, 116, 25880–25890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoriki, K.; Mori, T.; Kokabu, T.; Matsushima, H.; Umemura, S.; Tarumi, Y.; Kitawaki, J. Estrogen-related receptor alpha induces epithelial-mesenchymal transition through cancer-stromal interactions in endometrial cancer. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Du, P.; Ge, Z.; Jin, Y.; Ding, D.; Liu, X.; Zou, Q. TWIST1 and BMI1 in Cancer Metastasis and Chemoresistance. J. Cancer 2016, 7, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Lee, S.; Park, W.H.; Suh, D.H.; Kim, K.; Kim, Y.B.; No, J.H. Silencing Bmi1 expression suppresses cancer stemness and enhances chemosensitivity in endometrial cancer cells. Biomed. Pharmacother. 2018, 108, 584–589. [Google Scholar] [CrossRef]
- Wang, J.; Ai, Z.; Chen, J.; Teng, Y.; Zhu, J. Enhancer of zeste homolog 2 blockade by RNA interference is implicated with inhibited proliferation, invasion and promoted apoptosis in endometrial carcinoma. Oncol. Lett. 2018, 15, 9429–9435. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Wang, K.; Wang, J.; Qu, J.; Du, G.; Zhang, Y. SOX17 inhibits tumor metastasis via Wnt signaling in endometrial cancer. Onco Targets Ther. 2019, 12, 8275–8286. [Google Scholar] [CrossRef] [Green Version]
- Factors, A.; Kitson, S.J.; Rosser, M.; Fischer, D.P.; Marshall, K.M.; Clarke, R.B.; Crosbie, E.J. Targeting Endometrial Cancer Stem Cell Activity with Metformin Is Inhibited by Patient-Derived Adipocyte-Secreted Factors. Cancers 2019, 11, 653. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, X.; Zhao, L.; Zhou, J.; Cheng, Y.; Xu, B.; Wang, J.; Wei, L. Spontaneous formation of tumorigenic hybrids between human omental adipose-derived stromal cells and endometrial cancer cells increased motility and heterogeneity of cancer cells. Cell Cycle 2019, 18, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B.; et al. MiRNA Expression Profile of Human Subcutaneous Adipose and during Adipocyte Differentiation. PLoS ONE 2010, 5, e9022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Yue, Y.; Wang, R.; Zhang, Y.; Jin, Q.; Zhou, X. Overexpression of microRNA-194 suppresses the epithelial-mesenchymal transition in targeting stem cell transcription factor Sox3 in endometrial carcinoma stem cells. Tumor Biol. 2017, 39, 1010428317706217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhang, B.; Xu, N.; Wang, M.J.; Liu, Q. miR-326 regulates EMT and metastasis of endometrial cancer through targeting TWIST1. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3787–3793. [Google Scholar] [PubMed]
- Xiong, H.; Chen, R.; Liu, S.; Lin, Q.; Chen, H.; Jiang, Q. MicroRNA-183 induces epithelial-mesenchymal transition and promotes endometrial cancer cell migration and invasion in by targeting CPEB1. J. Cell. Biochem. 2018, 119, 8123–8137. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Sun, B.M.; Zhang, Y.Y.; Li, Y.J.; Huang, C.X.; Feng, F.Z.; Li, C. Upregulation of miR-183-5p is responsible for the promotion of apoptosis and inhibition of the epithelial-mesenchymal transition, proliferation, invasion and migration of human Endometrial cancer cells by downregulating Ezrin. Int. J. Mol. Med. 2018, 42, 2469–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Sui, D.; You, D.; Yao, S.; Zhang, L.; Wang, Y.; Zhao, J.; Zhang, Y. MiR-29a-5p inhibits proliferation and invasion and induces apoptosis in endometrial carcinoma via targeting TPX2. Cell Cycle 2018, 17, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Cai, Y.; An, R. miR-215 promotes epithelial to mesenchymal transition and proliferation by regulating LEFTY2 in endometrial cancer. Int. J. Mol. Med. 2018, 42, 1229–1236. [Google Scholar] [CrossRef]
- Deng, J.; Wang, W.; Yu, G.; Ma, X. MicroRNA-195 inhibits epithelial-mesenchymal transition by targeting G protein-coupled estrogen receptor 1 in endometrial carcinoma. Mol. Med. Rep. 2019, 20, 4023–4032. [Google Scholar] [CrossRef] [Green Version]
- Li, B.L.; Lu, W.; Qu, J.J.; Ye, L.; Du, G.Q.; Wan, X.P. Loss of exosomal miR-148b from cancer-associated fibroblasts promotes endometrial cancer cell invasion and cancer metastasis. J. Cell. Physiol. 2019, 234, 2943–2953. [Google Scholar] [CrossRef]
- Xie, P.; Cao, H.; Li, Y.; Wang, J.; Cui, Z. Knockdown of lncRNA CCAT2 inhibits endometrial cancer cells growth and metastasis via sponging miR-216b. Cancer Biomark. 2017, 21, 123–133. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Z.; Chen, W.; Zhai, W.; Pan, J.; Pang, H.; Li, X. H19 promotes endometrial cancer progression by modulating epithelial-mesenchymal transition. Oncol. Lett. 2017, 13, 363–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.A.; Kim, L.K.; Kim, Y.T.; Heo, T.H.; Kim, H.J. Long non-coding RNA steroid receptor activator promotes the progression of endometrial cancer via wnt/β-catenin signaling pathway. Int. J. Biol. Sci. 2020, 16, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, A.M.; Girolimetti, G.; Procaccini, M.; Marchio, L.; Livi, A.; Borghese, G.; Porcelli, A.M.; De Iaco, P.; Gasparre, G. Potential for mitochondrial DNA sequencing in the differential diagnosis of gynaecological malignancies. Int. J. Mol. Sci. 2018, 19, 2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chihara, N.; Amo, T.; Tokunaga, A.; Yuzuriha, R.; Wolf, A.M.; Asoh, S.; Suzuki, H.; Uchida, E.; Ohta, S. Mitochondrial DNA Alterations in Colorectal Cancer Cell Lines. J. Nippon Med. Sch. 2011, 78, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.B.; Ye, Y.; Huang, M.; Chang, D.W.; Kamat, A.M.; Pu, X.; Dinney, C.P.; Wu, X. Mitochondrial DNA Content as Risk Factor for Bladder Cancer and Its Association with Mitochondrial DNA Polymorphisms. Cancer Prev. Res. 2015, 8, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Kalsbeek, A.M.F.; Chan, E.K.F.; Corcoran, N.M.; Hovens, C.M.; Hayes, V.M. Mitochondrial genome variation and prostate cancer: A review of the mutational landscape and application to clinical management. Oncotarget 2017, 8, 71342–71357. [Google Scholar] [CrossRef] [Green Version]
- Witte, J.; Lehmann, S.; Wulfert, M.; Yang, Q.; Röher, H.D. Mitochondrial DNA Mutations in Differentiated Thyroid Cancer with Respect to the Age Factor. World J. Surg. 2007, 31, 51–59. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, V.W.S.; Xue, W.-C.; Tsang, P.C.K.; Cheung, A.N.Y.; Ngan, H.Y.S. The increase of mitochondrial DNA content in endometrial adenocarcinoma cells: A quantitative study using laser-captured microdissected tissues. Gynecol. Oncol. 2005, 98, 104–110. [Google Scholar] [CrossRef]
- Guerra, F.; Kurelac, I.; Magini, P.; Cormio, A.; Santini, D.; Ceccarelli, C.; Gasparre, G. Mitochondrial DNA genotyping reveals synchronous nature of simultaneously detected endometrial and ovarian cancers. Gynecol. Oncol. 2011, 122, 457–458. [Google Scholar] [CrossRef]
- Liu, V.W.S.; Yang, H.J.; Wang, Y.; Tsang, P.C.K.; Cheung, A.N.Y.; Chiu, P.M.; Ng, T.Y.; Wong, L.C.; Nagley, P.; Ngan, H.Y.S. High frequency of mitochondrial genome instability in human endometrial carcinomas. Br. J. Cancer 2003, 89, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.S.; Wang, Y.; Yang, H.-J.; Tsang, P.C.K.; Ng, T.-Y.; Wong, L.-C.; Nagley, P.; Ngan, H.Y.S. Mitochondrial DNA variant 16189T>C is associated with susceptibility to endometrial cancer. Hum. Mutat. 2003, 22, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hu, Y.; Chen, B.; Tang, W.; Han, X.; Yu, H.; Xiao, C. Mitochondrial polymorphisms as risk factors for endometrial cancer in southwest China. Int. J. Gynecol. Cancer 2006, 16, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wen, X.; Luan, F.; Fu, T.; Gao, C.; Du, H.; Guo, T.; Han, J.; Huangfu, L.; Cheng, X.; et al. EIF3B is associated with poor outcomes in gastric cancer patients and promotes cancer progression via the PI3K/AKT/mTOR signaling pathway. Cancer Manag. Res. 2019, 11, 7877–7891. [Google Scholar] [CrossRef] [Green Version]
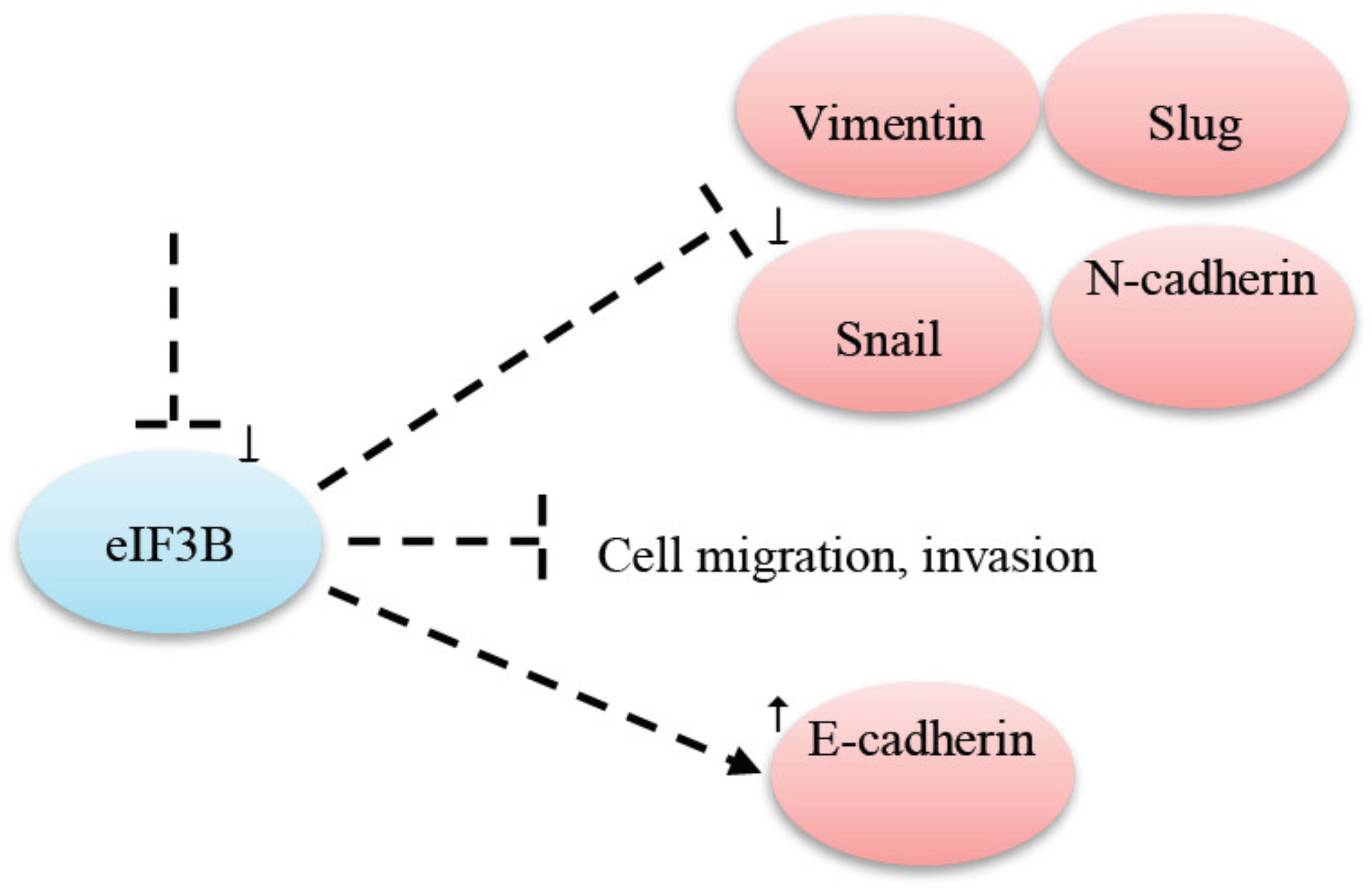
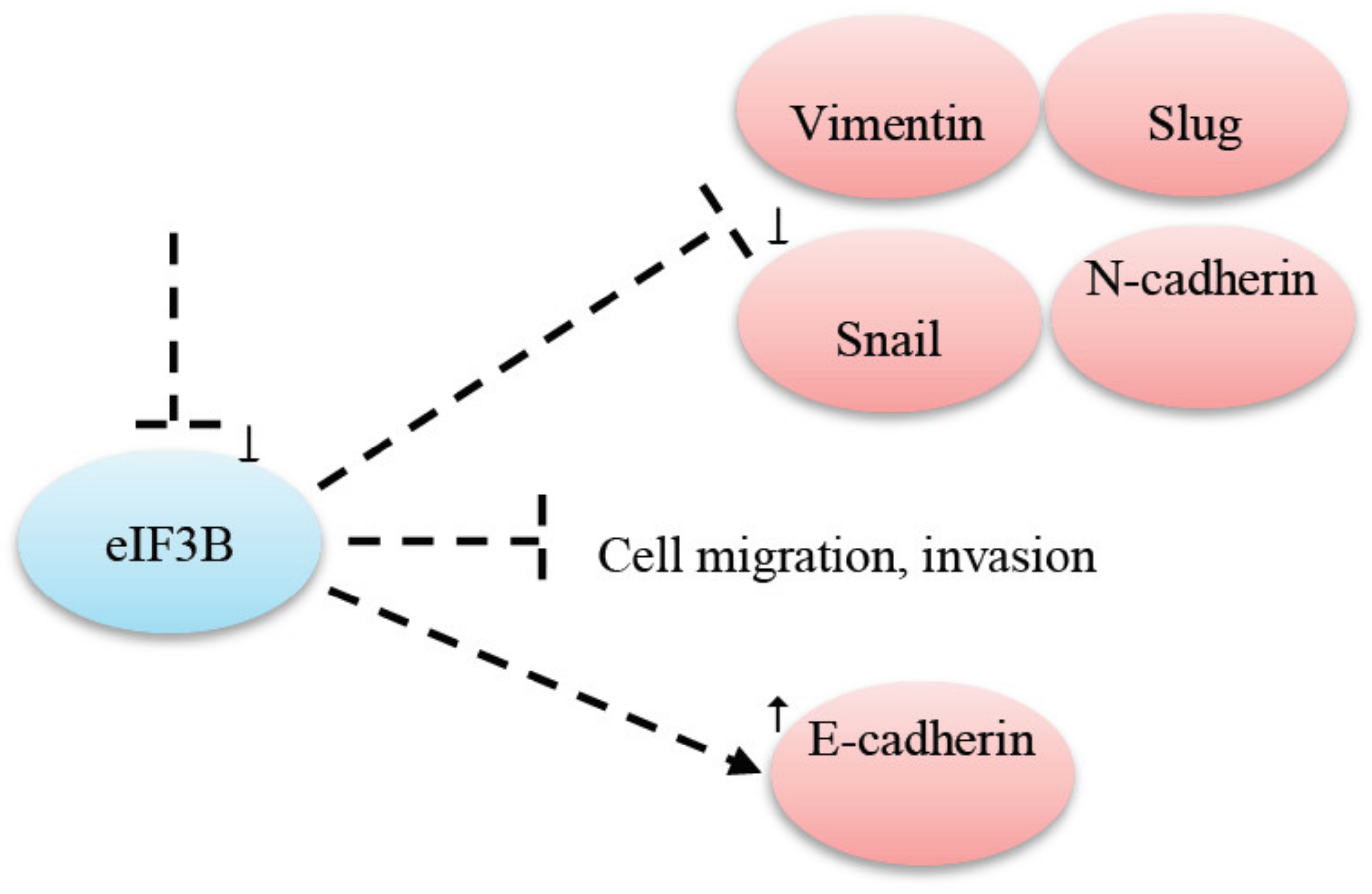
- Min, J.; Chen, H. Eukaryotic initiation factor 3B downregulation suppresses cell proliferation, migration and invasion while it induces cell apoptosis by blocking the β-catenin pathway in endometrial cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 3595–3603. [Google Scholar] [PubMed]
- Fang, L.; Gao, L.; Xie, L.; Xiao, G. Eukaryotic translation initiation factor 5A-2 involves in doxorubicin-induced epithelial-mesenchymal transition in oral squamous cell carcinoma cells. J. Cancer 2018, 9, 3479–3488. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.C.; Chu, T.Y.; Liu, H.W. Reciprocal crosstalk between endometrial carcinoma and mesenchymal stem cells via transforming growth factor-β/transforming growth factor receptor and C-X-C motif chemokine ligand 12/C-X-C chemokine receptor type 4 aggravates malignant phenotypes. Oncotarget 2017, 8, 115202–115214. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Attademo, L.; Scotto, G.; Genta, S.; Ghisoni, E.; Tuninetti, V.; Aglietta, M.; Pignata, S.; Valabrega, G. Endometrial cancer stem cells: Role, characterization and therapeutic implications. Cancers 2019, 11, 1820. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Chen, C.; Lin, A.; Xie, Y. Endoplasmic reticulum stress-mediated membrane expression of CRT/ERp57 induces immunogenic apoptosis in drug-resistant endometrial cancer cells. Oncotarget 2017, 8, 58754–58764. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| ClinicalTrials.Gov Identifier | Tumour | Drug | Purpose | Outcome |
|---|---|---|---|---|
| NCT01056757 | Breast cancer | Ribavirin (eIF4E-inhibitor) | Inclusion of patients with high eIF4E expression | Terminated (overlap with other ribavirin-study). |
| NCT01309490 | Malignant solid tumours | Ribavirin (eIF4E-inhibitor) | Inclusion of patients with high eIF4E expression | Recruiting |
| NCT01234025 | Prostate cancer (castration resistant) | ISIS EIF4E Rx (Antisense oligonucleotide against eIF4E) | Progression free survival following treatment with docetaxel and prednisolone, with/without ISI EIF4E Rx | Completed (no results posted) |
| NCT01234038 | Non-small cell lung cancer (stage IV) | ISIS EIF4E Rx (Antisense oligonucleotide against eIF4E) | Progression free survival following treatment with carboplatin and paclitaxel, with/without ISI EIF4E Rx | Completed (no results posted) |
| NCT00458549 | Prostate cancer | Omega-3 fatty acids | Determine whether neoadjuvant n-3 polyunsaturated fatty acids induce eIF2alpha-phosphorylation in prostate cancer patients | Terminated (slow accrual) |
| NCT00903708 | Advanced cancers | LY2275796 (antisense anti-cancer drug targeting eIF4E) | Pharmacokinetic and pharmacodynamic evaluation of intravenous LY2275796 | Completed (no results posted) |
| NCT04092673 | Advanced solid tumours | Zotatifin (= eFT226; inhibitor of eIF4A1-mediated translation) | Dose escalation and cohort-expansion study | Recruiting |
| NCT01268579 | Tonsil and/or base of tongue squamous cell carcinoma | Ribavirin (eIF4E-inhibitor) | Explore whether 2-week ribavirin therapy decreases tumour expression of eIF4E | Active, not recruiting |
| NCT01675128 | Advanced solid tumours, colorectal cancer | ISIS EIF4E Rx (Antisense oligonucleotide against eIF4E) | Pharmacokinetics and pharmacodynamics; investigate maximum tolerated dose and safety of ISIS EIF4E Rx in combination with irinotecan | Completed (Results published [32]) |
| NCT01056523 | Acute myeloid leukaemia | Ribavirin (eIF4E-inhibitor) | Pharmacokinetics, pharmacodynamics and efficacy of ribavirin in combination with cytarabine arabinoside | Completed (Results published [33]) |
| NCT02073838 | Acute myeloid leukaemia | Ribavirin (eIF4E-inhibitor) | Effect of ribavirin in combination with hedgehog-inhibitor vismodegib and/or cytidine analogue decitabine | Unknown (last status: recruiting) |
| MiR | Target Gene | Function |
|---|---|---|
| 183 [55] | CPEB1 | CPEB1 overexpression mediated 3′-UTR shortening and induces EMT proliferation, migration, invasion. |
| 183 [56] | Ezrin | Upregulation of miR-183 represses Ezrin and reduces metastatic potential. |
| 29 [57] | TPX2 | TPX2 overexpression enhances cell proliferation and invasion as well as enhancing apoptosis. |
| 215 [58] | LEFTY2 | Upregulation of miR-215 lead to LEFTY2 decrease and subsequent diminishment of mesenchymal to epithelial transition. |
| 326 [54] | TWIST1 | TWIST1 and miR-326 are directly correlated and miR-326 is downregulated in EC, leading to cell proliferation, migration, invasion and EMT. |
| 195 [59] | GPER | Overexpression leads to downregulation of MMP-2 and MMP-9 and decreased phosphorylation of PI3K and AKT. |
| 148 [60] | DNMT1 | Loss of expression in exomes leads to induction of EMT in CAFs. |
| 194 [53] | SOX3 | Upregulation of miR-194 leads to SOX3 suppression and decrease of stem cell invasion. |
| Eukaryotic Initiation Factors | Function |
|---|---|
| eIF3B | eIF3B knockdown prevented cell migration and invasion in gastric cancer [74]. |
| eIF2A-2 | eiF2A-2 has a direct involvement in the E-M transition. A knockdown led to an increase in E-cadherin expression and to a decrease in vimentin expression [76]. |
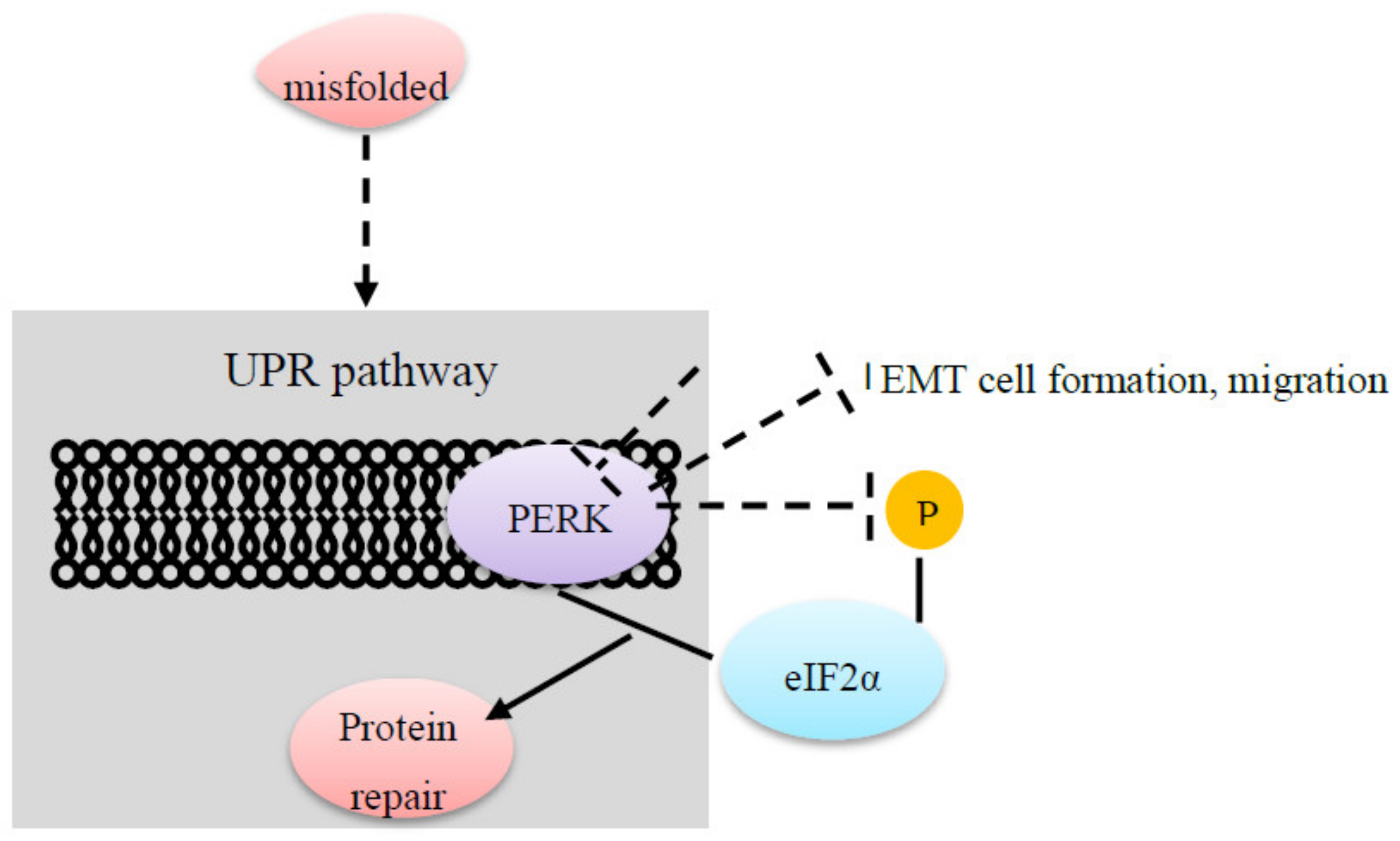
| eIF2alpha | PERK in the endoplasmic reticulum interacts with eIF2alpha for an integrated stress response to repair proteins. Inhibition of PERK impacts EIF2alpha phosphorylation [37]. PERK-eIF2alpha is involved in noncancerous processes and in carcinogenesis [37]. |
| eIF2beta | eIF2beta was found overexpressed in EC [25,26] and is related to a poorer patient survival [27]. |
| eIF3C, eIF3H | eIF3C and eIF3H as well as eIF2alpha, eIF4G, eIF5 and eIF6 were found aberrantly expressed in EC [22]. |
| eIF4E | miR-320a and miR-340-5p were downregulated in normal tissue [25]. eIF4E mRNA is regulated by miR-320a and miR-340-5p [25]. A downregulation respectively a phosphorylation of eIF4E led to an impairment of cellular migration affecting MMP-9 and MMP-3 [25]. TGF-beta1 is responsible for the phosphorylation of eIF4E and was suppressed by miR-320a and miR-340-5p and therefore resulted in an impairment of epithelial-to-mesenchymal transition [25]. eIF4E was found overexpressed in 50–60% of specimens [23,24]. A correlation with advanced cancer stage, lymph node metastasis and poor patient survival was found [23,24]. |
| eIF4G | eIF4G levels were found higher in type II EC compared to type I and led to a worse patient outcome [22]. |
| eIF6 | eIF6 was found downregulated in EC [22]. |
| eIF5 | eIF5 was found upregulated in EC [22]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobočan, M.; Smolle, M.A.; Schatz, C.; Haybaeck, J. The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer. Cancers 2020, 12, 2074. https://doi.org/10.3390/cancers12082074
Sobočan M, Smolle MA, Schatz C, Haybaeck J. The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer. Cancers. 2020; 12(8):2074. https://doi.org/10.3390/cancers12082074
Chicago/Turabian StyleSobočan, Monika, Maria Anna Smolle, Christoph Schatz, and Johannes Haybaeck. 2020. "The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer" Cancers 12, no. 8: 2074. https://doi.org/10.3390/cancers12082074
APA StyleSobočan, M., Smolle, M. A., Schatz, C., & Haybaeck, J. (2020). The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer. Cancers, 12(8), 2074. https://doi.org/10.3390/cancers12082074