YB-1 Knockdown Inhibits the Proliferation of Mesothelioma Cells through Multiple Mechanisms

 ,
,  , and
, and
Abstract
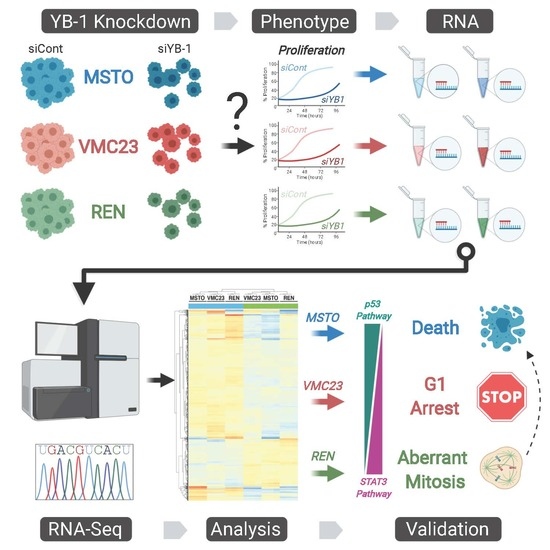
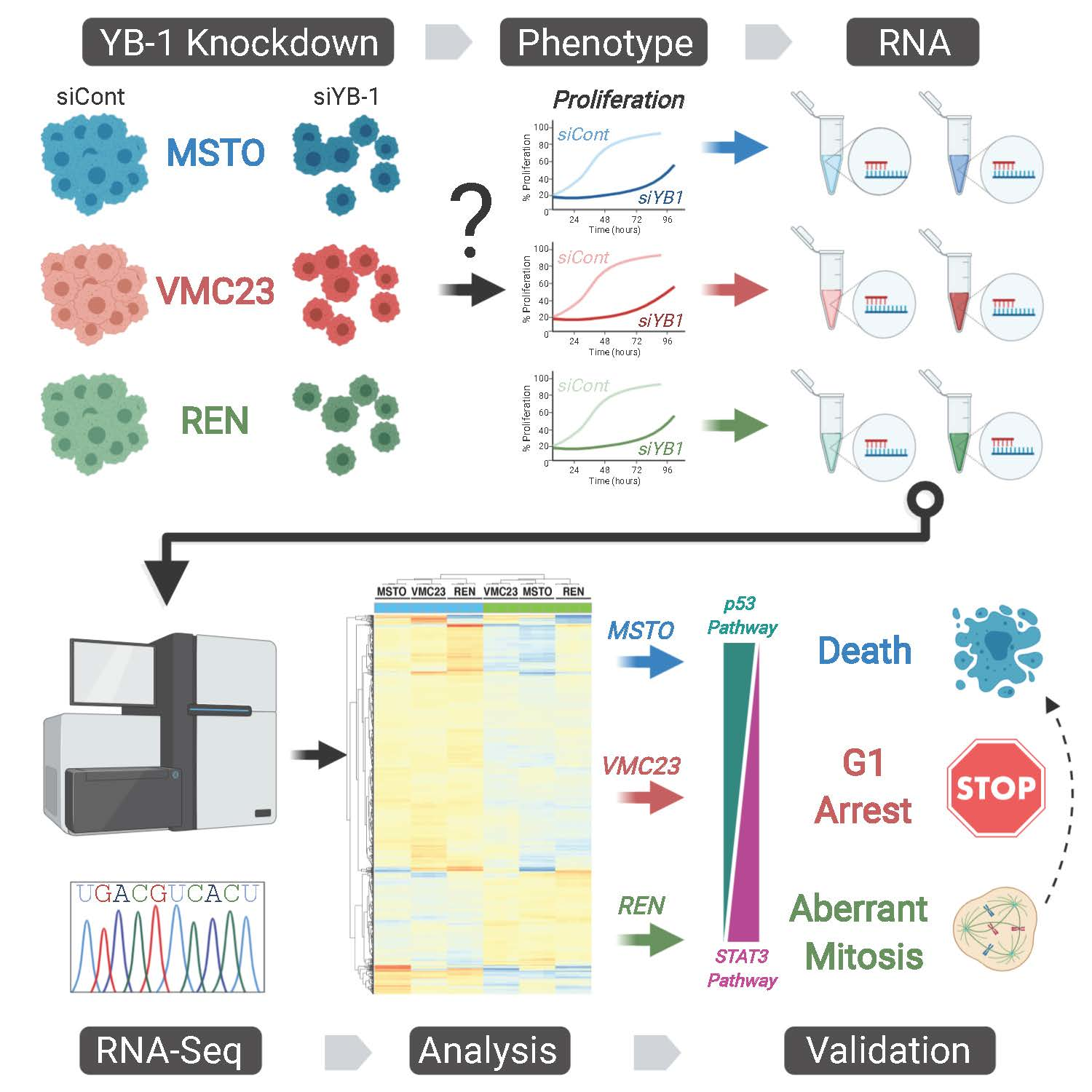{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
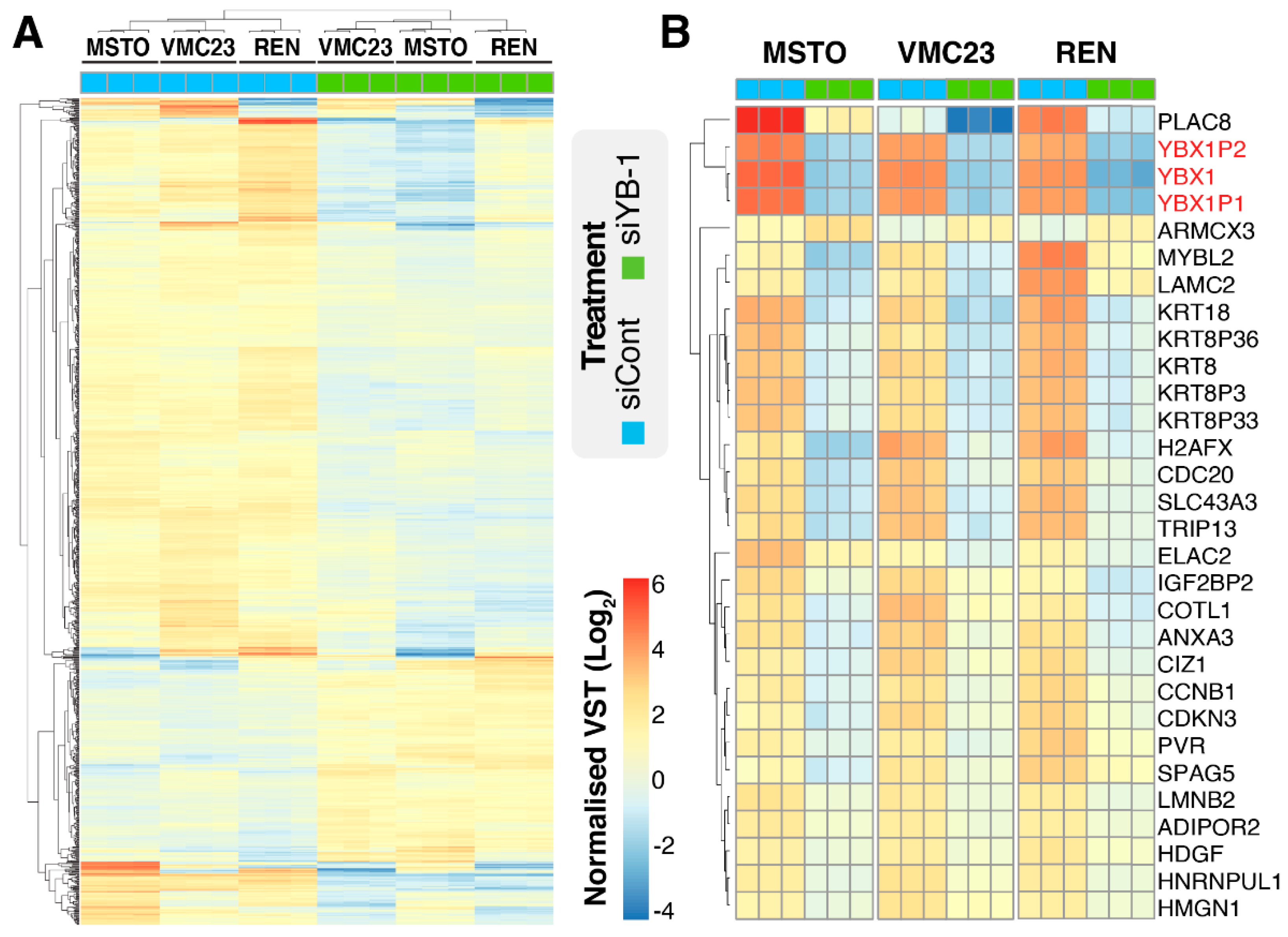
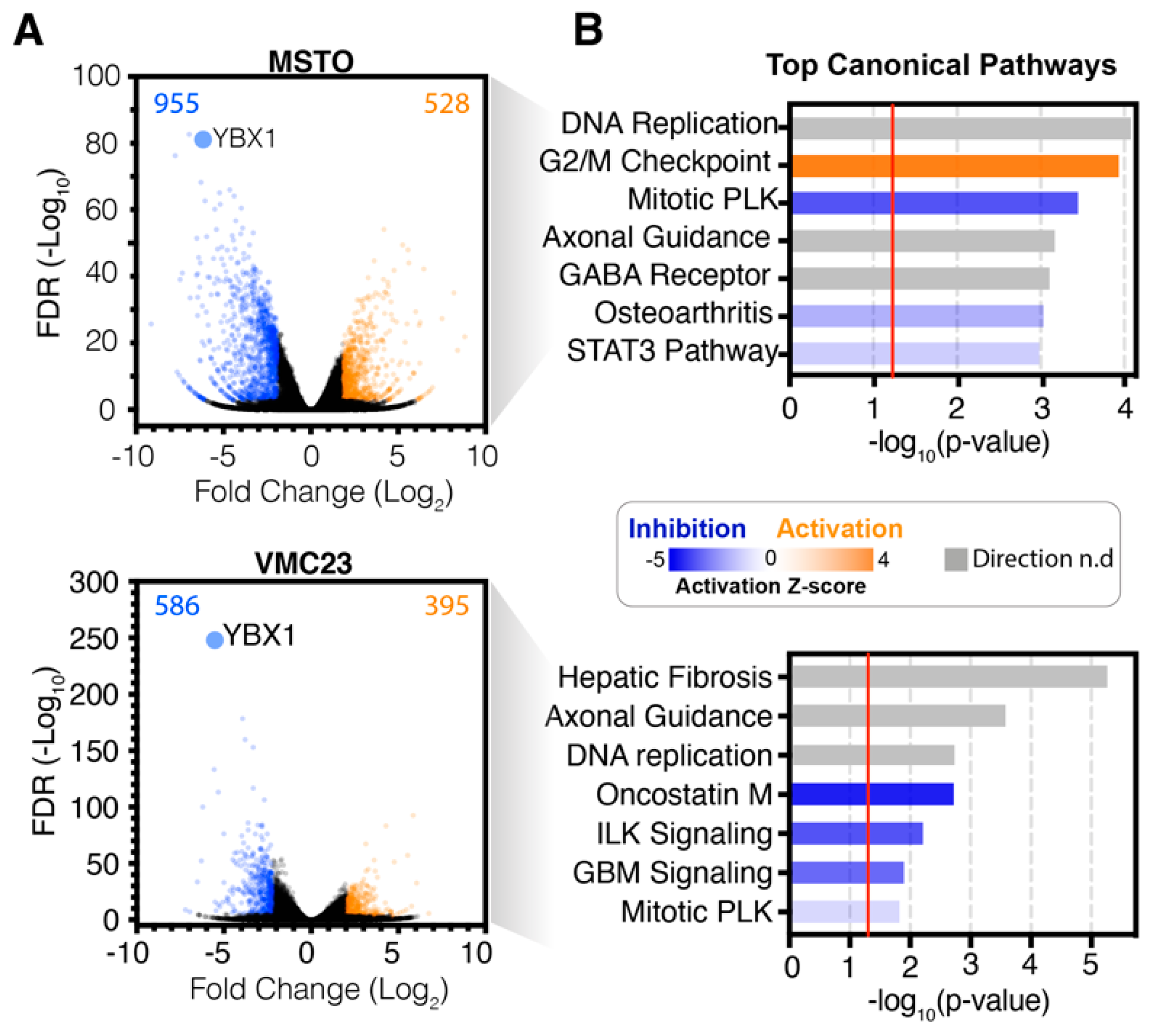
2.1. YB-1 Knockdown Alters Gene Transcription in MPM Cells
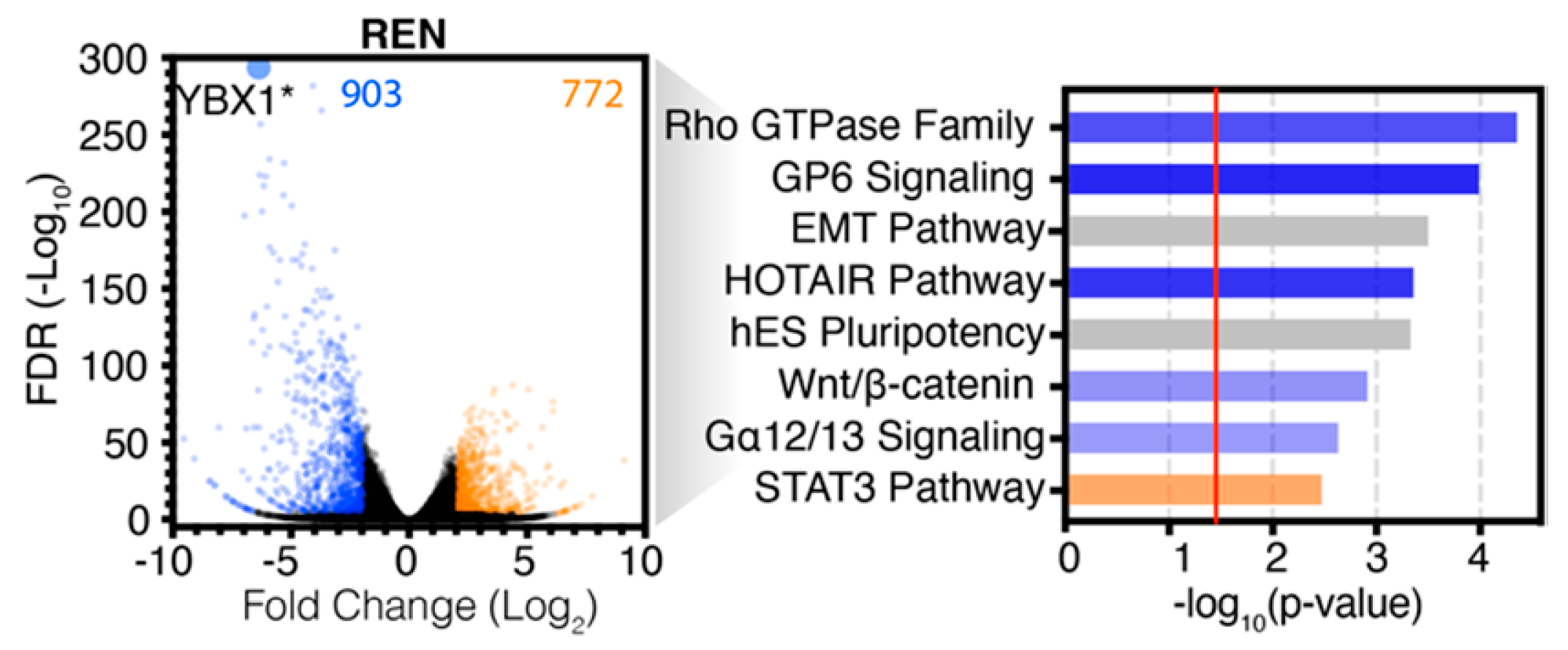
2.2. Analysis of RNA-seq Data Identifies Unique Enrichment of Canonical Pathways Following YB-1 Knockdown
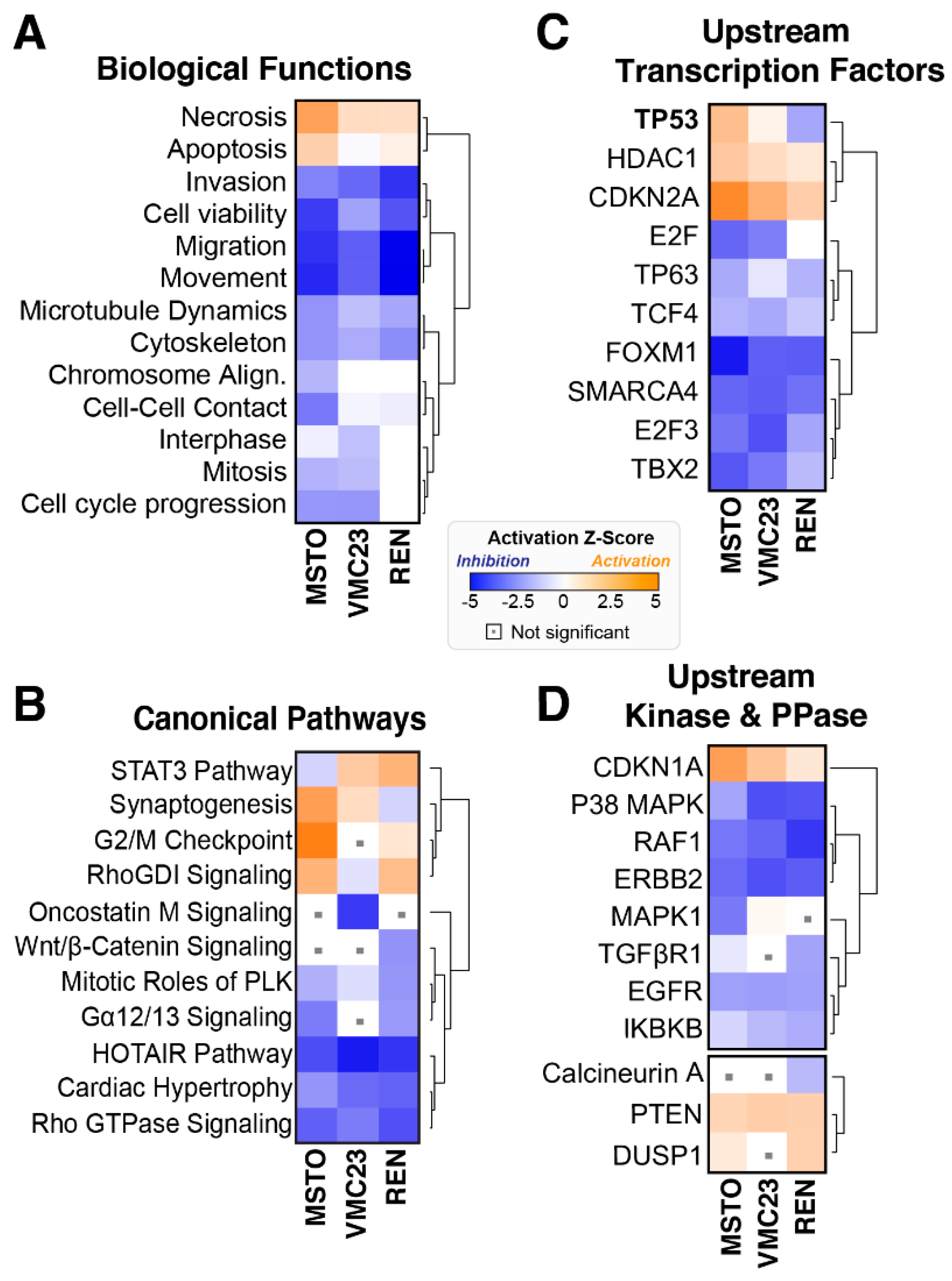
2.3. Comparative Analysis of RNA-seq Data Identifies Differential Response to YB-1 Inhibition in MPM Cells
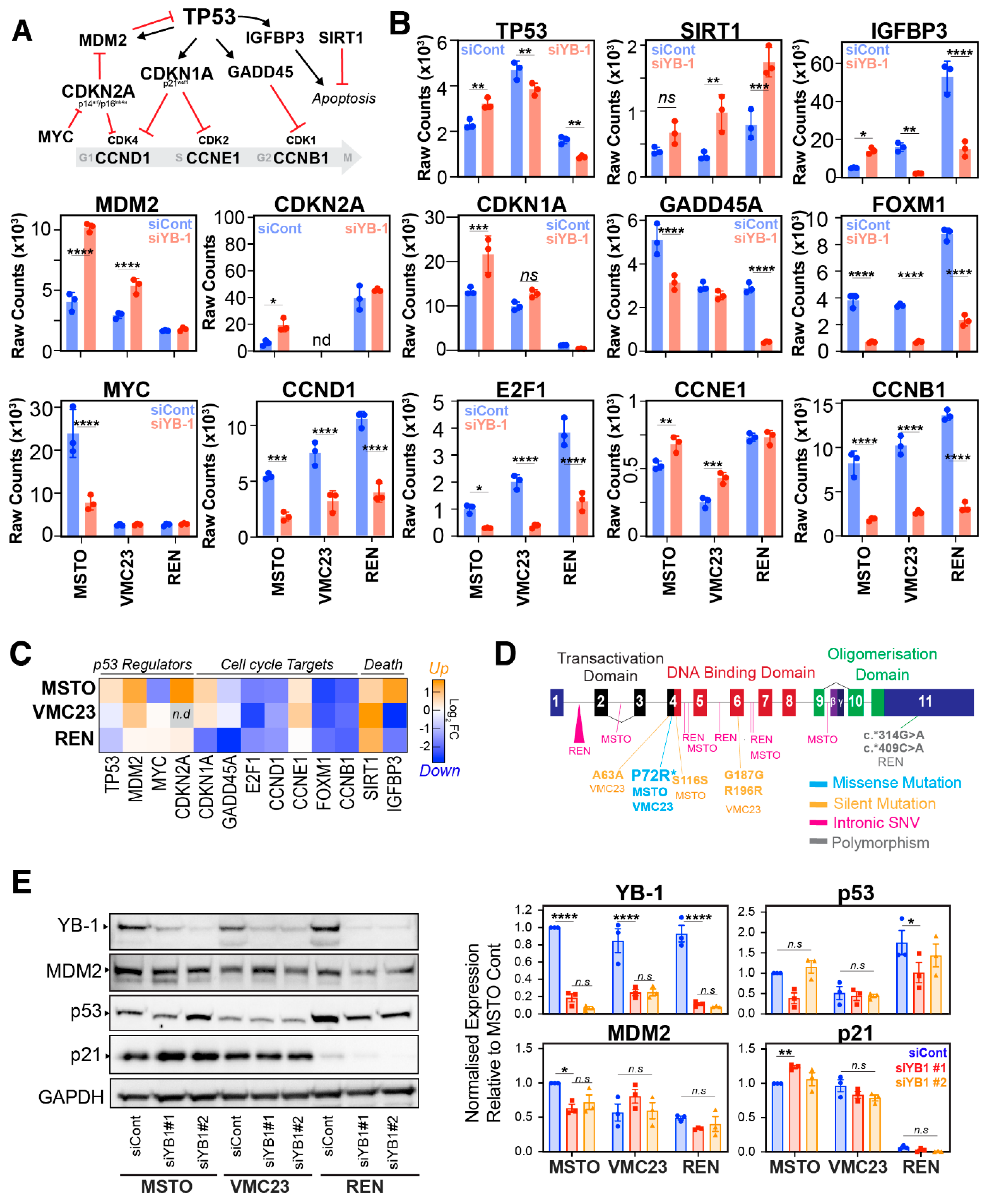
2.4. Differential p53 Signalling after YB-1 Knockdown in MPM Cells
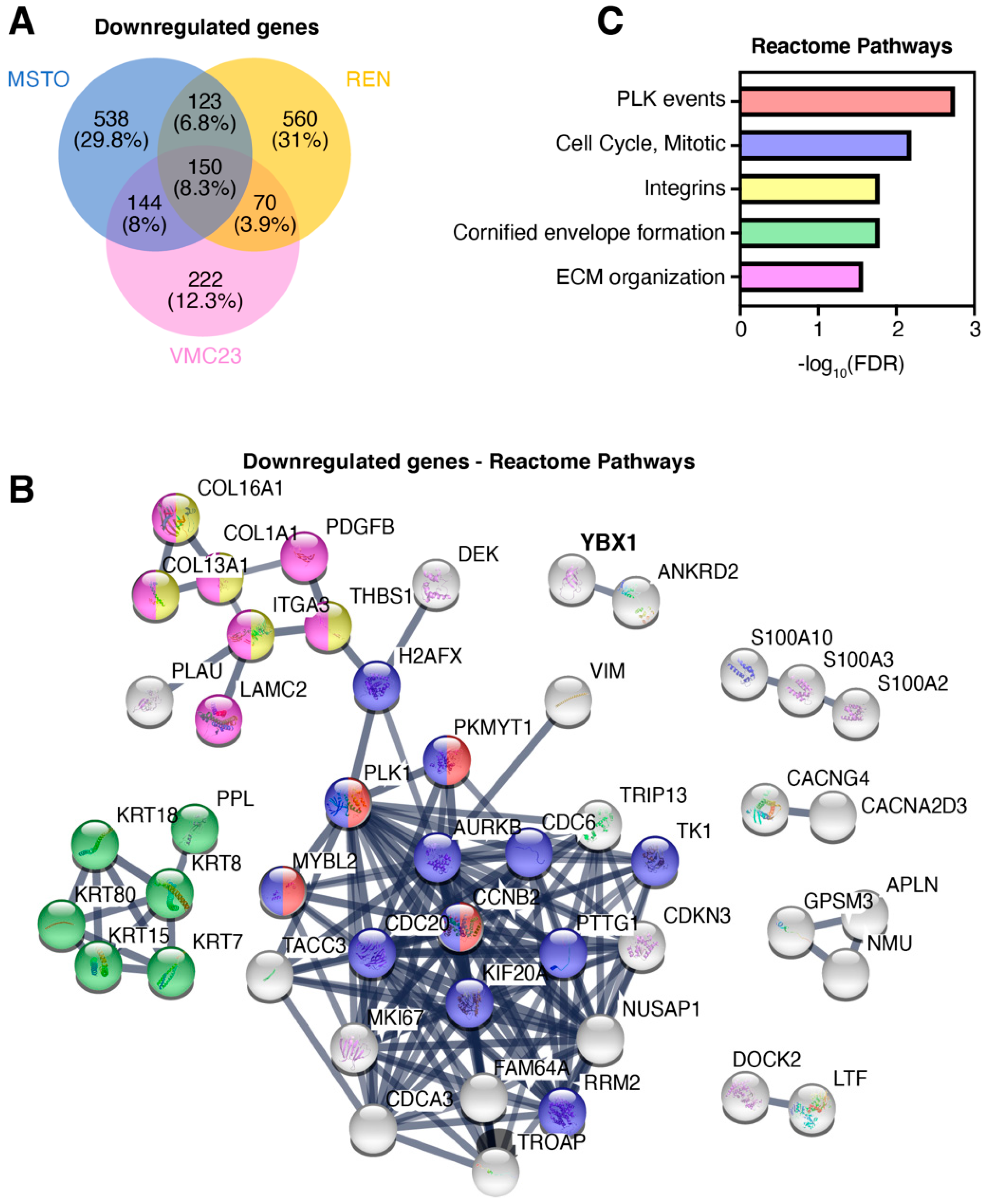
2.5. Identifying Potential Core Target Genes of YB-1
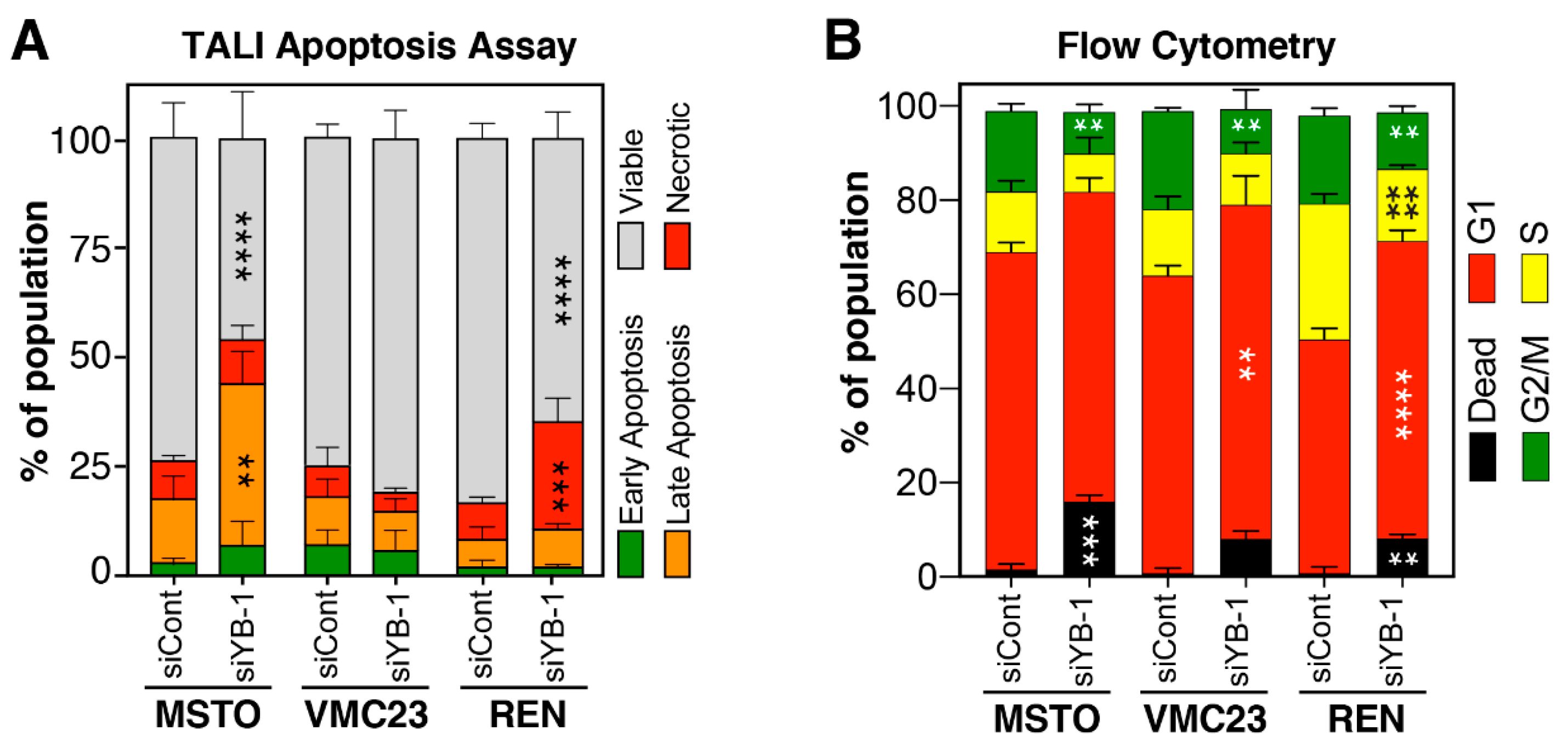
2.6. Growth Inhibition after YB-1 Knockdown Occurs via Apoptosis or an Altered Cell Cycle
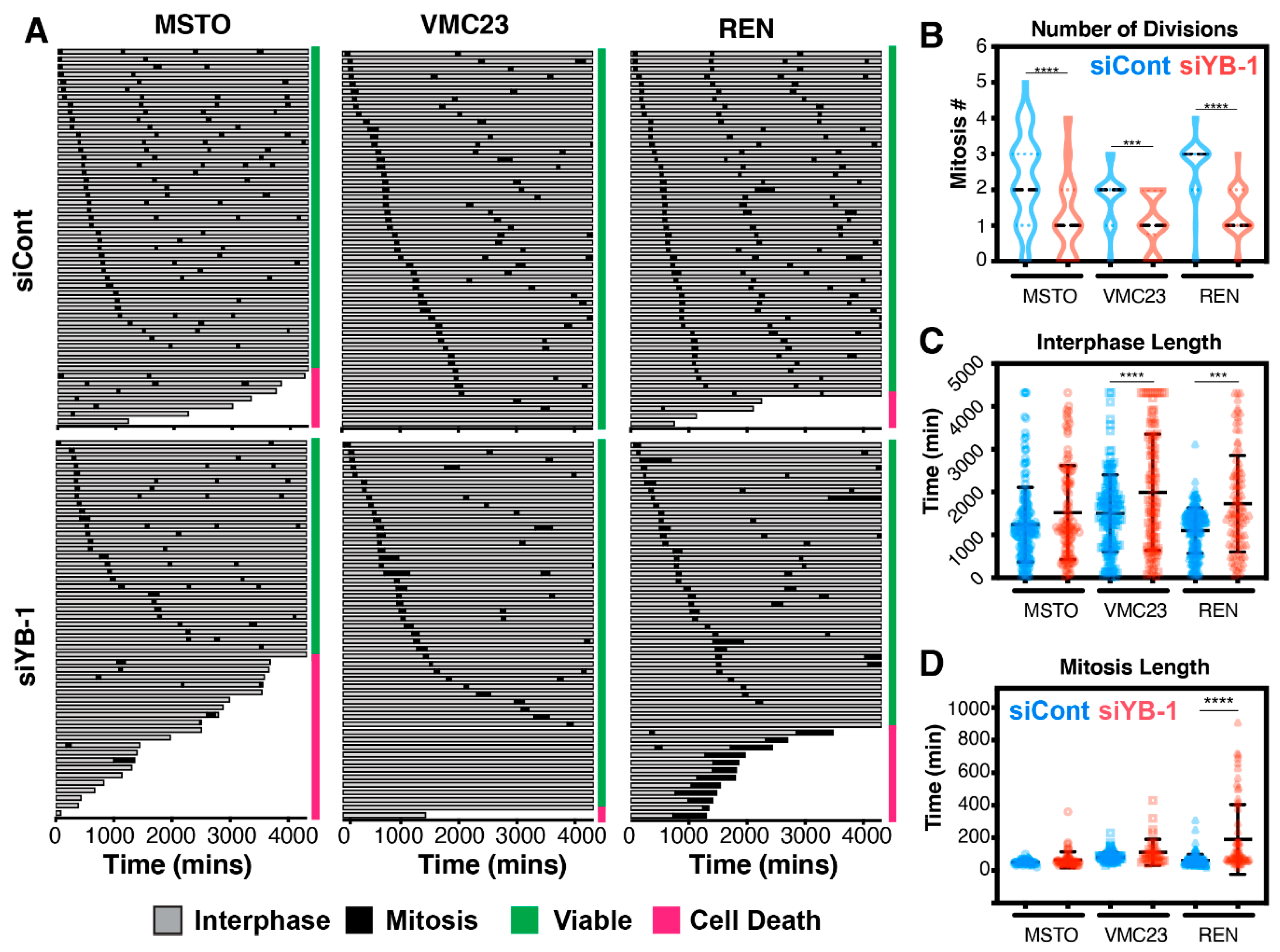
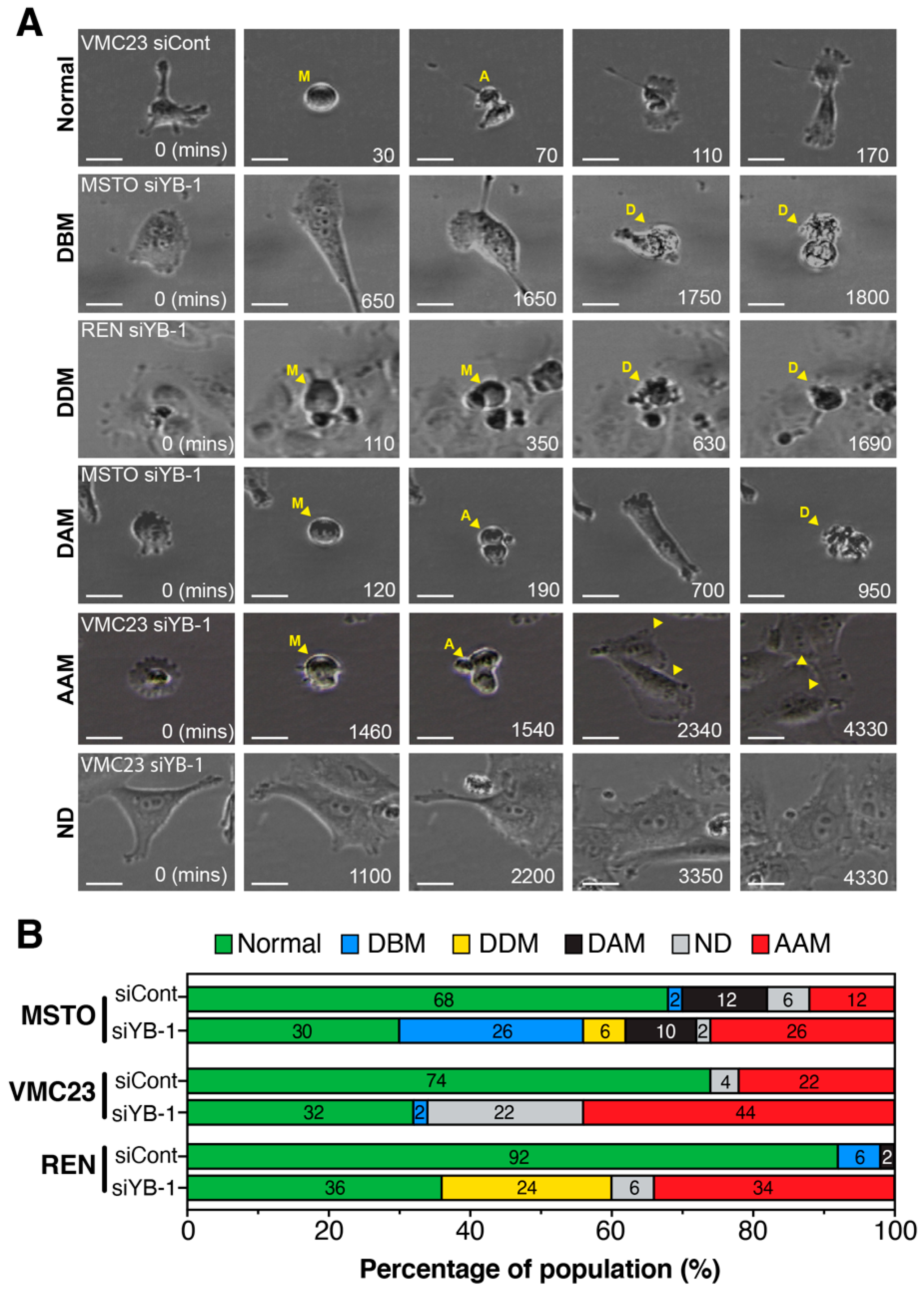
2.7. Live-Cell Imaging Confirms that YB-1 Knockdown Induces Three Different Growth Inhibitory Phenotypes
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection of siRNA
4.3. RNA Isolation, RNA Sequencing (RNA-seq), SNV Alignment and Analysis
4.4. Protein Isolation and Western Blot
4.5. SYBR Green-Based Cell Proliferation Assays
4.6. Apoptosis Assays
4.7. Cell Flow Cytometry Cell Cycle Analysis
4.8. Live-Cell Imaging and Cell Fate Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Australian Institute of Health & Welfare. Cancer Data in Australia; AIHW: Canberra, Australia, 2019. Available online: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/contents/summary (accessed on 20 July 2020).
- Purek, L.; Laroumagne, S.; Dutau, H.; Maldonado, F.; Astoul, P. Miliary mesothelioma: A new clinical and radiological presentation in mesothelioma patients with prolonged survival after trimodality therapy. J. Thorac. Oncol. 2011, 6, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, B.; Klikovits, T.; Hoda, M.; Lang, G.; Szirtes, I.; Setinek, U.; Rozsas, A.; Renyi-Vamos, F.; Laszlo, V.; Grusch, M. Ki67 index is an independent prognostic factor in epithelioid but not in non-epithelioid malignant pleural mesothelioma: A multicenter study. Br. J. Cancer 2015, 112, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Mutti, L.; Peikert, T.; Robinson, B.W.; Scherpereel, A.; Tsao, A.S.; de Perrot, M.; Woodard, G.A.; Jablons, D.M.; Wiens, J.; Hirsch, F.R. Scientific advances and new frontiers in mesothelioma therapeutics. J. Thorac. Oncol. 2018, 13, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Ventola, C.L. Cancer immunotherapy, part 3: Challenges and future trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- De Gooijer, C.J.; Borm, F.J.; Scherpereel, A.; Baas, P. Immunotherapy in Malignant Pleural Mesothelioma. Front. Oncol. 2020, 10, 187. [Google Scholar] [CrossRef]
- Sementino, E.; Menges, C.W.; Kadariya, Y.; Peri, S.; Xu, J.; Liu, Z.; Wilkes, R.G.; Cai, K.Q.; Rauscher III, F.J.; Klein-Szanto, A.J. Inactivation of Tp53 and Pten drives rapid development of pleural and peritoneal malignant mesotheliomas. J. Cell. Physiol. 2018, 233, 8952–8961. [Google Scholar]
- Ladanyi, M. Implications of P16/CDKN2A deletion in pleural mesotheliomas. Lung Cancer 2005, 49, S95–S98. [Google Scholar] [CrossRef]
- Agarwal, V.; Campbell, A.; Beaumont, K.L.; Cawkwell, L.; Lind, M.J. PTEN protein expression in malignant pleural mesothelioma. Tumor Biol. 2013, 34, 847–851. [Google Scholar] [CrossRef]
- Johnson, T.G.; Schelch, K.; Cheng, Y.Y.; Williams, M.; Sarun, K.H.; Kirschner, M.B.; Kao, S.; Linton, A.; Klebe, S.; McCaughan, B.C. Dysregulated expression of the MicroRNA miR-137 and its target YBX1 contribute to the invasive characteristics of malignant pleural mesothelioma. J. Thorac. Oncol. 2018, 13, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.G.; Schelch, K.; Mehta, S.; Burgess, A.; Reid, G. Why Be One Protein When You Can Affect Many? The Multiple Roles of YB-1 in Lung Cancer and Mesothelioma. Front. Cell Dev. Biol. 2019, 7, 221. [Google Scholar] [CrossRef]
- Mordovkina, D.; Lyabin, D.N.; Smolin, E.A.; Sogorina, E.M.; Ovchinnikov, L.P.; Eliseeva, I. Y-Box Binding Proteins in mRNP Assembly, Translation, and Stability Control. Biomolecules 2020, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. AACR 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Lasham, A.; Print, C.G.; Woolley, A.G.; Dunn, S.E.; Braithwaite, A.W. YB-1: Oncoprotein, prognostic marker and therapeutic target? Biochem. J. 2012, 449, 11–23. [Google Scholar] [CrossRef]
- Lasham, A.; Samuel, W.; Cao, H.; Patel, R.; Mehta, R.; Stern, J.L.; Reid, G.; Woolley, A.G.; Miller, L.D.; Black, M.A. YB-1, the E2F pathway, and regulation of tumor cell growth. J. Natl. Cancer Inst. 2011, 104, 133–146. [Google Scholar] [CrossRef]
- Harada, M.; Kotake, Y.; Ohhata, T.; Kitagawa, K.; Niida, H.; Matsuura, S.; Funai, K.; Sugimura, H.; Suda, T.; Kitagawa, M. YB-1 promotes transcription of cyclin D1 in human non-small-cell lung cancers. Genes Cells 2014, 19, 504–516. [Google Scholar] [CrossRef]
- Evdokimova, V.; Ruzanov, P.; Anglesio, M.S.; Sorokin, A.V.; Ovchinnikov, L.P.; Buckley, J.; Triche, T.J.; Sonenberg, N.; Sorensen, P.H. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol. Cell Biol. 2006, 26, 277–292. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Guo, T.; Yu, W.; Li, J.; Tang, Z.; Yu, Z.; Zhao, L.; Zhang, Y.; Wang, Z. YBX1 regulates tumor growth via CDC25a pathway in human lung adenocarcinoma. Oncotarget 2016, 7, 82139–82157. [Google Scholar]
- Stratford, A.L.; Habibi, G.; Astanehe, A.; Jiang, H.; Hu, K.; Park, E.; Shadeo, A.; Buys, T.P.; Lam, W.; Pugh, T. Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 2007, 9, R61. [Google Scholar] [CrossRef]
- Bommert, K.; Effenberger, M.; Leich, E.; Küspert, M.; Murphy, D.; Langer, C.; Moll, R.; Janz, S.; Mottok, A.; Weissbach, S. The feed-forward loop between YB-1 and MYC is essential for multiple myeloma cell survival. Leukemia 2013, 27, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Izumi, H.; Imamura, T.; Takano, H.; Ise, T.; Uchiumi, T.; Kuwano, M.; Kohno, K. Direct interaction of p53 with the Y-box binding protein, YB-1: A mechanism for regulation of human gene expression. Oncogene 2000, 19, 6194–6202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lasham, A.; Moloney, S.; Hale, T.; Homer, C.; Zhang, Y.F.; Murison, J.G.; Braithwaite, A.W.; Watson, J. The Y-box-binding protein, YB1, is a potential negative regulator of the p53 tumor suppressor. J. Biol. Chem. 2003, 278, 35516–35523. [Google Scholar] [CrossRef] [PubMed]
- Homer, C.; Knight, D.A.; Hananeia, L.; Sheard, P.; Risk, J.; Lasham, A.; Royds, J.A.; Braithwaite, A.W. Y-box factor YB1 controls p53 apoptotic function. Oncogene 2005, 24, 8314–8325. [Google Scholar] [CrossRef]
- Tong, H.; Zhao, K.; Zhang, J.; Zhu, J.; Xiao, J. YB-1 modulates the drug resistance of glioma cells by activation of MDM2/p53 pathway. Drug Des. Dev. Ther. 2019, 13, 317–326. [Google Scholar] [CrossRef]
- Fujii, T.; Seki, N.; Namoto-Matsubayashi, R.; Takahashi, H.; Inoue, Y.; Toh, U.; Kage, M.; Shirouzu, K. YB-1 prevents apoptosis via the mTOR/STAT3 pathway in HER-2-overexpressing breast cancer cells. Future Oncol. 2009, 5. [Google Scholar] [CrossRef]
- Takeuchi, A.; Shiota, M.; Tatsugami, K.; Yokomizo, A.; Kuroiwa, K.; Dejima, T.; Tanaka, S.; Itsumi, M.; Eto, M.; Naito, S. YB-1 suppression induces STAT3 proteolysis and sensitizes renal cancer to interferon-α. Cancer Immunol. Immunother. 2013, 62, 517–527. [Google Scholar] [CrossRef]
- Alemasova, E.; Lavrik, O. At the interface of three nucleic acids: The role of RNA-binding proteins and poly (ADP-ribose) in DNA repair. Acta Nat. 2017, 9, 4–16. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Moor, N.A.; Naumenko, K.N.; Kutuzov, M.M.; Sukhanova, M.V.; Pestryakov, P.E.; Lavrik, O.I. Y-box-binding protein 1 as a non-canonical factor of base excision repair. Biochim. Et Biophys. Acta (Bba)-Proteins Proteom. 2016, 1864, 1631–1640. [Google Scholar] [CrossRef]
- Ise, T.; Nagatani, G.; Imamura, T.; Kato, K.; Takano, H.; Nomoto, M.; Izumi, H.; Ohmori, H.; Okamoto, T.; Ohga, T. Transcription factor Y-box binding protein 1 binds preferentially to cisplatin-modified DNA and interacts with proliferating cell nuclear antigen. Cancer Res. 1999, 59, 342–346. [Google Scholar]
- Gaudreault, I.; Guay, D.; Lebel, M. YB-1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cisplatin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucleic Acids Res. 2004, 32, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Ying, X.; Zhou, J.; Chen, Y.; Luo, X.; Xie, S.; Wang, Q.C.; Hu, W.; Wang, L. The novel KLF4/PLAC8 signaling pathway regulates lung cancer growth. Cell Death Dis. 2018, 9, 603. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Vuong, J.; Marzec, K.A.; Nicolai de Lichtenberg, U.; O’Donoghue, S.I.; Jensen, L.J. SnapShot: S-Phase Entry and Exit. Cell 2019, 179, 802. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Song, X.Q.; Cai, J.P.; Zhang, S. HOTAIR: A cancer-related long non-coding RNA. Neoplasma 2014, 61, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, H.; Li, Y.; Wang, R.; Li, Y.; Zhang, H.; Ren, D.; Liu, H.; Kang, C.; Chen, J. HOTAIR, a long noncoding RNA, is a marker of abnormal cell cycle regulation in lung cancer. Cancer Sci. 2018, 109, 2717–2733. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, J.T.; Baek, K.E.; Kim, B.Y.; Lee, H.G. Regulation of Rho GTPases by RhoGDIs in Human Cancers. Cells 2019, 8, 1037. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Grimberg, A. P53 and IGFBP-3: Apoptosis and cancer protection. Mol. Genet. Metab. 2000, 70, 85–98. [Google Scholar] [CrossRef]
- Marzec, K.A.; Lin, M.Z.; Martin, J.L.; Baxter, R.C. Involvement of p53 in insulin-like growth factor binding protein-3 regulation in the breast cancer cell response to DNA damage. Oncotarget 2015, 6, 26583–26598. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.; Todorova, K.; Wang, J.; Horos, R.; Lee, K.K.; Neel, V.A.; Negri, G.L.; Sorensen, P.H.; Lee, S.W.; Hentze, M.W.; et al. The RNA-binding protein YBX1 regulates epidermal progenitors at a posttranscriptional level. Nat. Commun. 2018, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, Z.; Liu, H.; Zhang, Q.; Cai, X.; Qu, Y.; Xu, M.; Lu, L. Y-box Protein-1 Regulates the Expression of Collagen I in Hepatic Progenitor Cells via PDGFR-beta/ERK/p90RSK Signalling. Stem Cells Int. 2017, 2017, 6193106. [Google Scholar] [CrossRef]
- Caldon, C.E.; Burgess, A. Label free, quantitative single-cell fate tracking of time-lapse movies. MethodsX 2019, 6, 2468–2475. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.P.; Nair, S.; Shyamasundar, S.; Chua, P.J.; Muniasamy, U.; Matsumoto, K.; Gunaratne, J.; Bay, B.H. Silencing Y-box binding protein-1 inhibits triple-negative breast cancer cell invasiveness via regulation of MMP1 and beta-catenin expression. Cancer Lett. 2019, 452, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Nagasu, S.; Sudo, T.; Kinugasa, T.; Yomoda, T.; Fujiyoshi, K.; Shigaki, T.; Akagi, Y. Y-box-binding protein 1 inhibits apoptosis and upregulates EGFR in colon cancer. Oncol. Rep. 2019, 41, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, K.; Nakano, K.; Cui, L.; Mizuuchi, Y.; Onishi, H.; Oda, Y.; Obika, S.; Tanaka, M.; Katano, M. Nuclear expression of Y-box binding protein-1 is associated with poor prognosis in patients with pancreatic cancer and its knockdown inhibits tumor growth and metastasis in mice tumor models. Int. J. Cancer 2016, 139, 433–445. [Google Scholar]
- Bader, A.G.; Felts, K.A.; Jiang, N.; Chang, H.W.; Vogt, P.K. Y box-binding protein 1 induces resistance to oncogenic transformation by the phosphatidylinositol 3-kinase pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 12384–12389. [Google Scholar]
- Cao, X.; Zhu, N.; Zhang, Y.; Chen, Y.; Zhang, J.; Li, J.; Hao, P.; Gao, C.; Li, L. Y-box protein 1 promotes hypoxia/reoxygenation-or ischemia/reperfusion-induced cardiomyocyte apoptosis via SHP-1-dependent STAT3 inactivation. J. Cell. Physiol. 2020, 1–12. [Google Scholar] [CrossRef]
- Basaki, Y.; Taguchi, K.; Izumi, H.; Murakami, Y.; Kubo, T.; Hosoi, F.; Watari, K.; Nakano, K.; Kawaguchi, H.; Ohno, S.; et al. Y-box binding protein-1 (YB-1) promotes cell cycle progression through CDC6-dependent pathway in human cancer cells. Eur. J. Cancer 2010, 46, 954–965. [Google Scholar] [CrossRef]
- Lyabin, D.N.; Eliseeva, I.A.; Smolin, E.A.; Doronin, A.N.; Budkina, K.S.; Kulakovskiy, I.V.; Ovchinnikov, L.P. YB-3 substitutes YB-1 in global mRNA binding. RNA Biol. 2020, 17, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of Stat3 in regulating p53 expression and function. Mol. Cell Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O.; Gascan, H. Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J. Biol Chem. 2000, 275, 18794–18800. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Khaku, S.; Jennis, M.; Zhou, Y.; Murphy, M.E. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol. Cancer Res. 2015, 13, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C., 3rd; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Samuels, Y.; Sullivan, A.; Zvelebil, M.; Breyssens, H.; Bisso, A.; Del Sal, G.; Syed, N.; Smith, P.; Gasco, M.; et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 2006, 38, 1133–1141. [Google Scholar] [CrossRef]
- Azzam, G.; Wang, X.; Bell, D.; Murphy, M.E. CSF1 is a novel p53 target gene whose protein product functions in a feed-forward manner to suppress apoptosis and enhance p53-mediated growth arrest. PLoS ONE 2013, 8, e74297. [Google Scholar] [CrossRef]
- Dutertre, M.; Sanchez, G.; De Cian, M.C.; Barbier, J.; Dardenne, E.; Gratadou, L.; Dujardin, G.; Le Jossic-Corcos, C.; Corcos, L.; Auboeuf, D. Cotranscriptional exon skipping in the genotoxic stress response. Nat. Struct Mol. Biol. 2010, 17, 1358–1366. [Google Scholar] [CrossRef]
- Mehta, S.; Algie, M.; Al-Jabri, T.; McKinney, C.; Kannan, S.; Verma, C.S.; Ma, W.; Zhang, J.; Bartolec, T.K.; Masamsetti, V.P. Critical role for cold shock protein YB-1 in cytokinesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Davies, A.H.; Barrett, I.; Pambid, M.R.; Hu, K.; Stratford, A.L.; Freeman, S.; Berquin, I.M.; Pelech, S.; Hieter, P.; Maxwell, C. YB-1 evokes susceptibility to cancer through cytokinesis failure, mitotic dysfunction and HER2 amplification. Oncogene 2011, 30, 3649–3660. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawaguchi, A.; Asaka, M.N.; Matsumoto, K.; Nagata, K. Centrosome maturation requires YB-1 to regulate dynamic instability of microtubules for nucleus reassembly. Sci. Rep. 2015, 5, 8768. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 5 February 2020).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, T.G.; Schelch, K.; Lai, K.; Marzec, K.A.; Kennerson, M.; Grusch, M.; Reid, G.; Burgess, A. YB-1 Knockdown Inhibits the Proliferation of Mesothelioma Cells through Multiple Mechanisms. Cancers 2020, 12, 2285. https://doi.org/10.3390/cancers12082285
Johnson TG, Schelch K, Lai K, Marzec KA, Kennerson M, Grusch M, Reid G, Burgess A. YB-1 Knockdown Inhibits the Proliferation of Mesothelioma Cells through Multiple Mechanisms. Cancers. 2020; 12(8):2285. https://doi.org/10.3390/cancers12082285
Chicago/Turabian StyleJohnson, Thomas G., Karin Schelch, Kaitao Lai, Kamila A. Marzec, Marina Kennerson, Michael Grusch, Glen Reid, and Andrew Burgess. 2020. "YB-1 Knockdown Inhibits the Proliferation of Mesothelioma Cells through Multiple Mechanisms" Cancers 12, no. 8: 2285. https://doi.org/10.3390/cancers12082285
APA StyleJohnson, T. G., Schelch, K., Lai, K., Marzec, K. A., Kennerson, M., Grusch, M., Reid, G., & Burgess, A. (2020). YB-1 Knockdown Inhibits the Proliferation of Mesothelioma Cells through Multiple Mechanisms. Cancers, 12(8), 2285. https://doi.org/10.3390/cancers12082285