PI3K p110α Blockade Enhances Anti-Tumor Efficacy of Abemaciclib in Human Colorectal Cancer Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
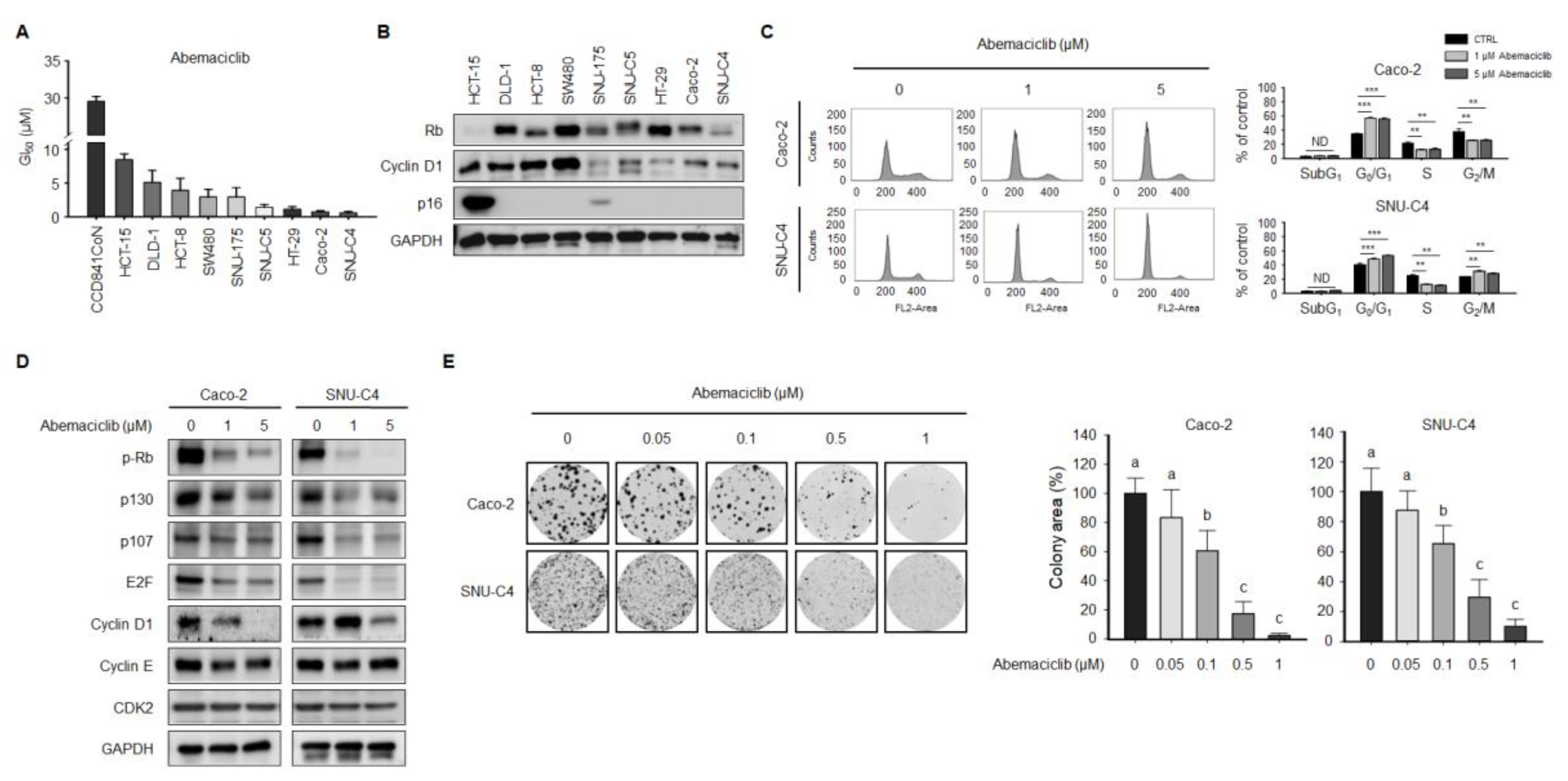
2.1. Abemaciclib Differentially Regulates Cell Proliferation Depending on Cyclin D1 and p16 Expression in Colon Cancer Cell Lines
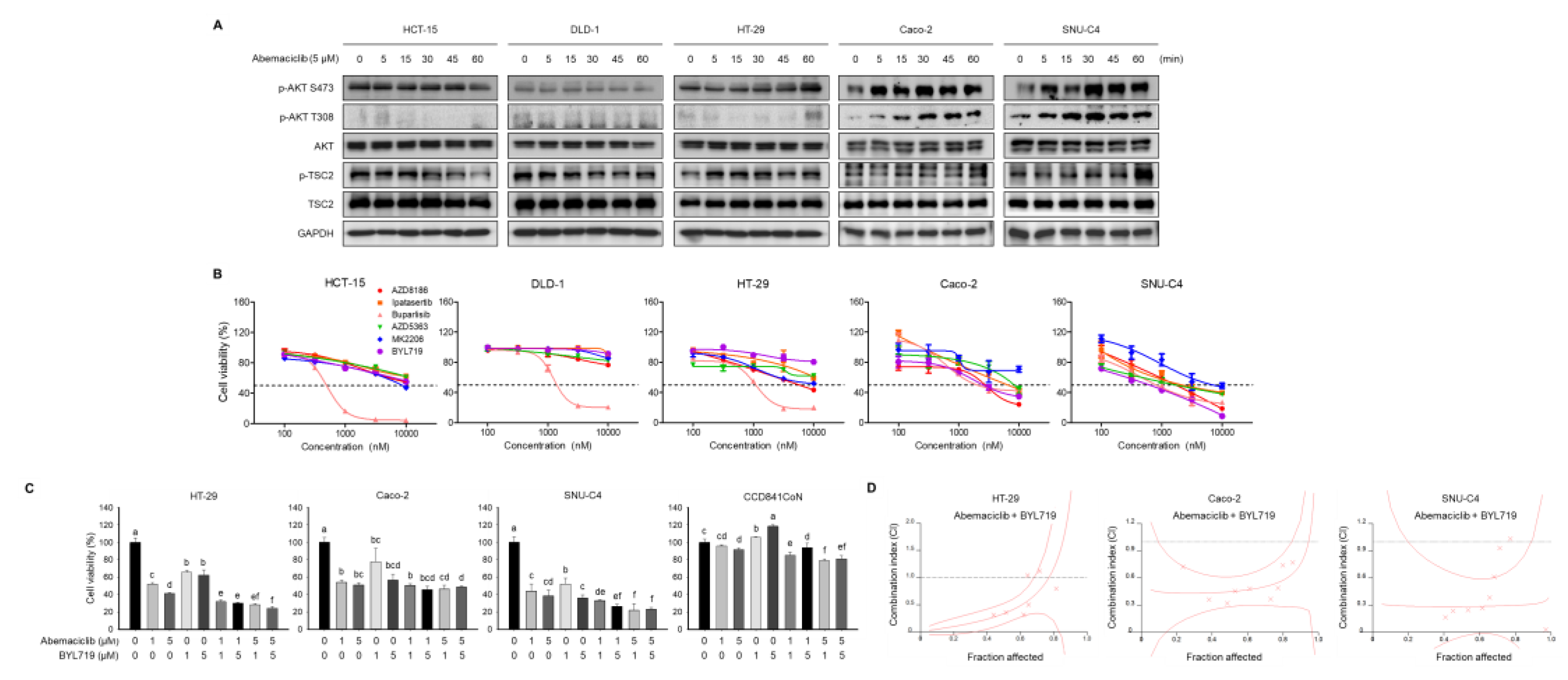
2.2. Abemaciclib and BYL719 Combination Shows Additive Anti-Proliferative Activity in PIK3CA Mutated Cell Lines
2.3. Abemaciclib and BYL719 Combination Induces G0/G1 Phase Arrest and Alters Regulatory Protein Expression in Caco-2 and SNU-C4 Cell Lines
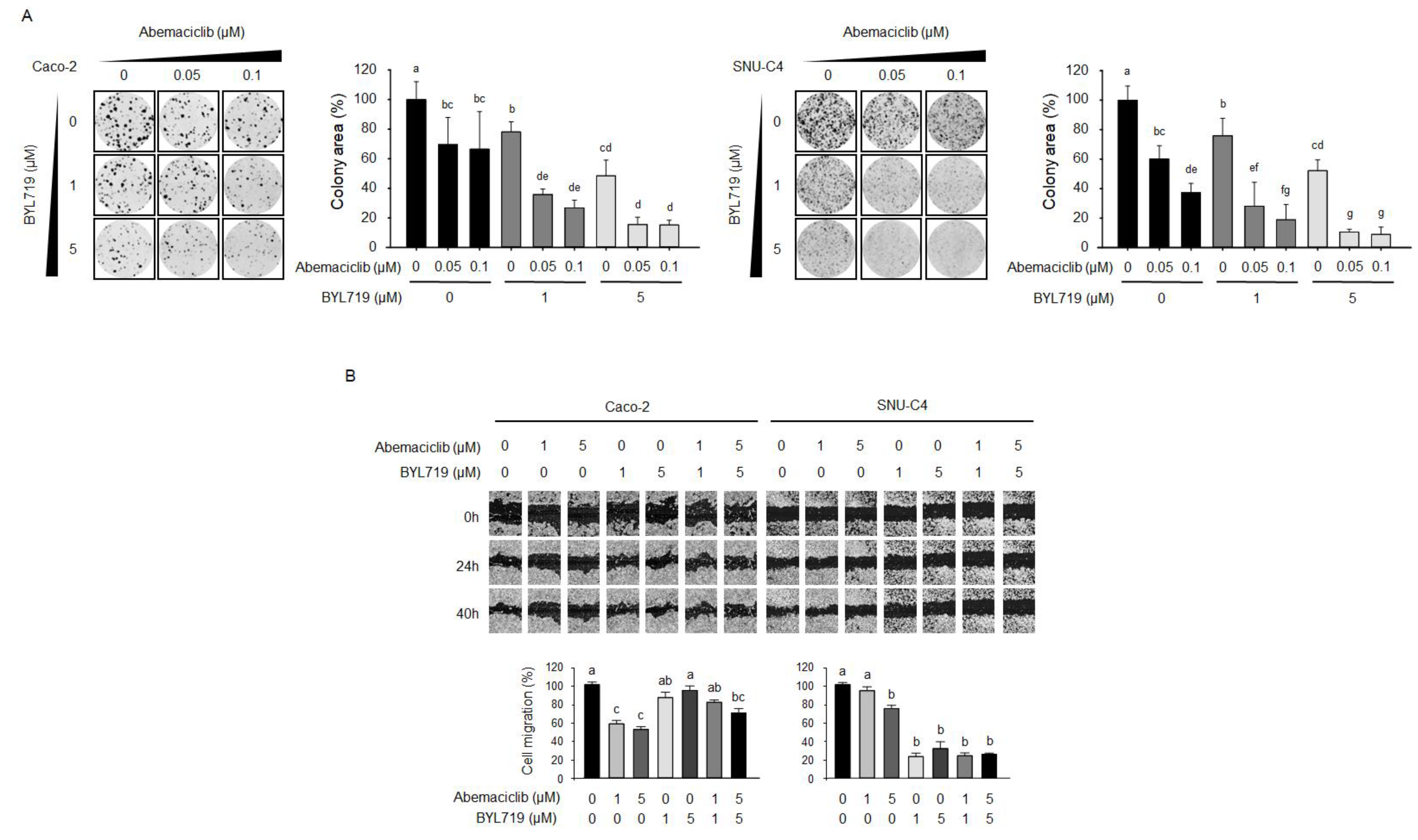
2.4. Abemaciclib and BYL719 Combination Inhibits Colony-Forming Activity and Cell Migration
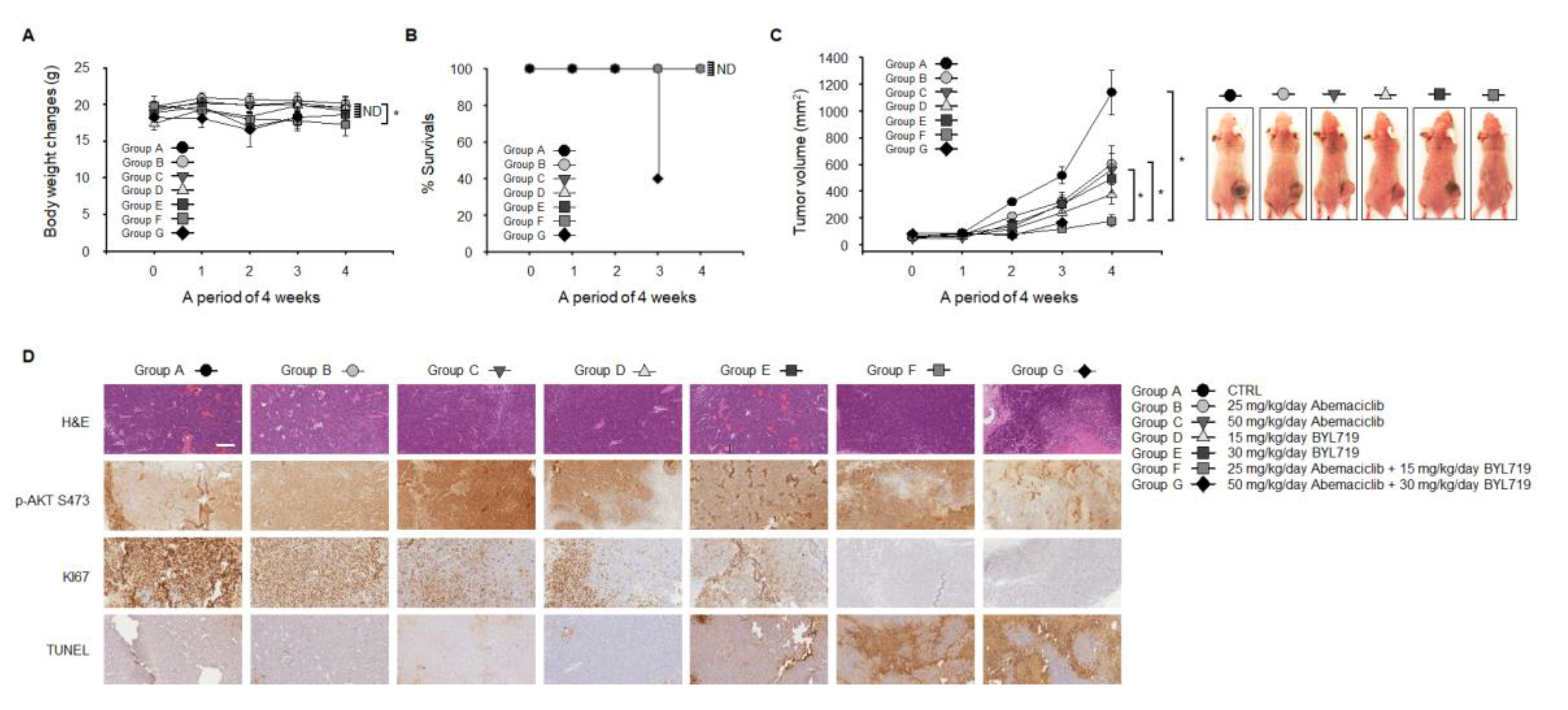
2.5. Abemaciclib and BYL719 Combination Inhibits Tumor Growth and Induces Apoptosis In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Colony-Formimg Assay
4.5. Cell Migration
4.6. Cell Cycle Analysis
4.7. Combination Index Analysis
4.8. Western Blot Analysis
4.9. Mouse Xenograft
4.10. Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jung, K.-W.; Won, Y.-J.; Kong, H.-J.; Lee, E.S. Cancer Statistics in Korea: Incidence, Mortality, Survival, and Prevalence in 2016. Cancer Res. Treat 2019, 51, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA. Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Harlow, E. The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef]
- Toncheva, D.; Petrova, D.; Tzenova, V.; Dimova, I.; Yankova, R.; Yordanov, V.; Damjanov, D.; Todorov, T.; Zaharieva, B. Tissue microarray analysis of cyclin D1 gene amplification and gain in colorectal carcinomas. Tumour Biol. 2004, 25, 157–160. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.A.; Douglas, J.J.; Ross, V.G.; Curran, S.; Murray, G.I.; Cassidy, J.; McLeod, H.L. Cyclin D1 protein expression and gene polymorphism in colorectal cancer. Aberdeen Colorectal Initiative. Int. J. Cancer 2000, 88, 77–81. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.-C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Filmus, J.; Robles, A.I.; Shi, W.; Wong, M.J.; Colombo, L.L.; Conti, C.J. Induction of cyclin D1 overexpression by activated ras. Oncogene 1994, 9, 3627–3633. [Google Scholar]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharek, S.J.; Xiong, Y.; Shumway, S.D. Negative Regulation of TSC1-TSC2 by Mammalian D-Type Cyclins. Cancer Res. 2005, 65, 11354–11360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res 2016, 22, 5696–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New. Drugs. 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Scheicher, R.; Hoelbl-Kovacic, A.; Bellutti, F.; Tigan, A.S.; Prchal-Murphy, M.; Heller, G.; Schneckenleithner, C.; Salazar-Roa, M.; Zöchbauer-Müller, S.; Zuber, J.; et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood 2015, 125, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [Green Version]
- Cancer Cell Line Encyclopedia. Available online: https://portals.broadinstitute.org/ccle (accessed on 4 April 2020).
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Wiedemeyer, W.R.; Dunn, I.F.; Quayle, S.N.; Zhang, J.; Chheda, M.G.; Dunn, G.P.; Zhuang, L.; Rosenbluh, J.; Chen, S.; Xiao, Y.; et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl. Acad. Sci. USA 2010, 107, 11501–11506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.; Jiang, Y.; Rugo, H.; Moulder, S.L.; Im, S.A.; Gelmon, K.A.; Dieras, V.; Martin, M.; Joy, A.A.; Toi, M.; et al. Biomarker analyses from the phase 3 PALOMA-2 trial of palbociclib (P) with letrozole (L) compared with placebo (PLB) plus L in postmenopausal women with ER + /HER2– advancedbreast cancer (ABC). Ann. Oncol. 2016, 27, vi554. [Google Scholar] [CrossRef]
- Donjerkovic, D.; Scott, D.W. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000, 10, 1–16. [Google Scholar] [CrossRef]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, T.; Xiao, Y.; Ong, C.; Daemen, A.; Friedman, L. Abstract P3-04-24: Identification of preclinical mechanisms driving acquired resistance to selective ERa degraders (SERDs), CDK4/6 inhibitors, or to combinations of both agents. Cancer Res. 2017, 77, 4–24. [Google Scholar] [CrossRef]
- Ligresti, G.; Militello, L.; Steelman, L.S.; Cavallaro, A.; Basile, F.; Nicoletti, F.; Stivala, F.; McCubrey, J.A.; Libra, M. PIK3CA mutations in human solid tumors: Role in sensitivity to various therapeutic approaches. Prog. Cell Cycle Res. 2009, 8, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Ihle, N.T.; Lemos, R., Jr.; Wipf, P.; Yacoub, A.; Mitchell, C.; Siwak, D.; Mills, G.B.; Dent, P.; Kirkpatrick, D.L.; Powis, G. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009, 69, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Nakamura, M.; Tokuda, E.; Niwa, T.; Ishida, T.; Hayashi, S.-I. Abstract P6-04-02: CDK6 might be a key factor for efficacy of CDK4/6 inhibitor and the hormone sensitivity following acquired resistance. Cancer Res. 2018, 78, 6-04-02. [Google Scholar] [CrossRef]
- O’Brien, N.; Conklin, D.; Beckmann, R.; Luo, T.; Chau, K.; Thomas, J.; Mc Nulty, A.; Marchal, C.; Kalous, O.; von Euw, E.; et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol. Cancer Ther. 2018, 17, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xu, K.; Liu, P.; Geng, Y.; Wang, B.; Gan, W.; Guo, J.; Wu, F.; Chin, Y.R.; Berrios, C.; et al. Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol. Cell 2016, 62, 929–942. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer. 2017, 3, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Forero-Torres, A.; Han, H.; Dees, E.C.; Wesolowski, R.; Bardia, A.; Kabos, P.; Layman, R.M.; Lu, J.M.; Kern, K.A.; Perea, R.; et al. Phase Ib study of gedatolisib in combination with palbociclib and endocrine therapy (ET) in women with estrogen receptor (ER) positive (+) metastatic breast cancer (MBC) (B2151009). J. Clin. Oncol. 2018, 36, 1040. [Google Scholar] [CrossRef]
- Pascual, J.; MacPherson, I.R.; Armstrong, A.C.; Ward, S.E.; Parmar, M.; Turner, A.J.; Bye, H.; Proszek, P.; Dodson, A.; Garcia-Murillas, I.; et al. PIPA: A phase Ib study of β-isoform sparing phosphatidylinositol 3-kinase (PI3K) inhibitor taselisib (T) plus palbociclib (P) and fulvestrant (FUL) in PIK3CA-mutant (mt) ER-positive and taselisib (T) plus palbociclib (P) in PIK3CA-mutant (mt) ER-negative advanced breast cancer. J. Clin. Oncol. 2019, 37, 1051. [Google Scholar] [CrossRef]
- Munster, P.N.; Hamilton, E.P.; Franklin, C.; Bhansali, S.; Wan, K.; Hewes, B.; Juric, D. Phase lb study of LEE011 and BYL719 in combination with letrozole in estrogen receptor-positive, HER2-negative breast cancer (ER+, HER2− BC). J. Clin. Oncol. 2014, 32, 533. [Google Scholar] [CrossRef]
- Mizrachi, A.; Dunn, L.; Ho, A.; Scaltriti, M. Abstract 61: Overcoming acquired resistance to PI3K inhibitors in head and neck squamous cell carcinoma with combination treatment. Cancer Res. 2017, 23, 61. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Kim, K.-J.; Sung, J.H.; Nam, M.; Suh, K.J.; Kim, J.-W.; Kim, S.H.; Kim, J.W.; Kim, Y.J.; Lee, K.-W.; et al. PI3K p110α Blockade Enhances Anti-Tumor Efficacy of Abemaciclib in Human Colorectal Cancer Cells. Cancers 2020, 12, 2500. https://doi.org/10.3390/cancers12092500
Lee HJ, Kim K-J, Sung JH, Nam M, Suh KJ, Kim J-W, Kim SH, Kim JW, Kim YJ, Lee K-W, et al. PI3K p110α Blockade Enhances Anti-Tumor Efficacy of Abemaciclib in Human Colorectal Cancer Cells. Cancers. 2020; 12(9):2500. https://doi.org/10.3390/cancers12092500
Chicago/Turabian StyleLee, Hyun Jung, Kui-Jin Kim, Ji Hea Sung, Milang Nam, Koung Jin Suh, Ji-Won Kim, Se Hyun Kim, Jin Won Kim, Yu Jung Kim, Keun-Wook Lee, and et al. 2020. "PI3K p110α Blockade Enhances Anti-Tumor Efficacy of Abemaciclib in Human Colorectal Cancer Cells" Cancers 12, no. 9: 2500. https://doi.org/10.3390/cancers12092500