EVI1 in Leukemia and Solid Tumors


Department of Cancer Biology and Genetics, College of Medicine, The Ohio State University, Columbus, OH 43210, USA
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(9), 2667; https://doi.org/10.3390/cancers12092667
Submission received: 8 July 2020
/
Revised: 2 September 2020
/
Accepted: 13 September 2020
/
Published: 18 September 2020
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Ecotropic viral integration site 1 (EVI1) is transcriptionally activated in a subset of myeloid leukemias. Since its discovery, other isoforms of EVI1 have been identified. It has been shown that EVI1 and its isoforms mainly function as transcription factors and to play important roles not only in leukemia but also in a variety of solid tumors. To provide a comprehensive understanding of this family of proteins, we summarize the currently available knowledge of expression and function of EVI1 and its isoforms in leukemia and solid tumors and provide insights of future studies.
Abstract
The EVI1 gene encodes for a transcription factor with two zinc finger domains and is transcriptionally activated in a subset of myeloid leukemias. In leukemia, the transcriptional activation of EVI1 usually results from chromosomal rearrangements. Besides leukemia, EVI1 has also been linked to solid tumors including breast cancer, lung cancer, ovarian cancer and colon cancer. The MDS1/EVI1 gene is encoded by the same locus as EVI1. While EVI1 functions as a transcription repressor, MDS1/EVI1 acts as a transcription activator. The fusion protein encoded by the AML1/MDS1/EVI1 chimeric gene, resulting from chromosomal translocations in a subset of chronic myeloid leukemia, exhibits a similar function to EVI1. EVI1 has been shown to regulate cell proliferation, differentiation and apoptosis, whereas the functions of MDS1/EVI1 and AML1/MDS1/EVI1 remain elusive. In this review, we summarize the genetic structures, biochemical properties and biological functions of these proteins in cancer.
1. Introduction
Ecotropic viral integration site 1 (EVI1) is an oncogenic transcription factor that plays an important role in development and oncogenesis. EVI1 normally exists as a single gene, or as a longer form in a fusion gene with myelodysplastic syndrome 1 (MDS1), called MDS1/EVI1 [1]. In a subset of chronic myeloid leukemia (CML) associated with t(3;21), EVI1 also exists as a chimeric fusion gene called AML1/MDS1/EVI1 [2,3].
Studies have shown that both EVI1 and AML1/MDS1/EVI1 mainly function as a transcription repressor, whereas MDS1/EVI1 functions as a transcription activator [4,5,6,7]. EVI1 is overexpressed in about 10% of adult acute myeloid leukemia (AML) and acts as a marker in myeloid malignancies for an aggressive disease with poor survival [8,9,10,11,12,13,14,15,16,17]. EVI1, MDS1/EVI1 and AML1/MDS1/EVI1 promote leukemogenesis by modulating differentiation, apoptosis, cell cycle and proliferation. EVI1 expression is not only upregulated in leukemia, but also in a number of solid tumors, including colorectal cancer, breast cancer, prostate cancer and ovarian cancer [18,19,20,21,22,23,24,25,26]. EVI1 has been shown to be redistributed or overexpressed in several types of solid tumors. Higher levels of EVI1 messenger RNA (mRNA) were detected in ovarian carcinoma compared to a normal ovary [25]. Similarly, EVI1 also exhibits a redistribution of expression in prostate cancer. In healthy prostatic tissues, EVI1 is expressed in the prostate stem cell compartment located at the basal layer. However, in a prostate cancer progression cohort consisting of 219 samples from patients with primary prostate cancer, lymph nodes and distant metastases, EVI1 was found to be heterogeneously distributed within samples [26]. Expression of EVI1 is low in adult normal small intestine and colon. However, previous studies on EVI1 in colorectal cancer have revealed that expression of EVI1 was upregulated in colon cancer patient samples and colon cancer cell lines, such as HCT116 cells and HT-29 cells [20,21]. Functionally, EVI1 modulates cell proliferation, cell cycle progression, migration and apoptosis. Several studies have shown that a number of signaling pathways are regulated by EVI1, including the epithelial-mesenchymal transition (EMT)-related, phosphatase and tensin homolog (PTEN)/AKT/the mammalian target of rapamycin (mTOR), transforming growth factor (TGF)-β signaling, apoptosis and cell cycle-related pathways [27,28,29,30,31,32,33,34,35]. Taken together, these studies suggest that upregulation of EVI1 expression could be a functionally important molecular event during cancer development and progression, and therefore EVI1 could be a potential molecular target for cancer treatment.
1.1. Structure of EVI1 and MDS1/EVI1
EVI1 was initially identified as a common ecotropic retrovirus integration site in murine leukemia [36]. Subsequent sequencing studies showed that EVI1 contains two DNA binding domains with seven and three repeats of the zinc finger motif, respectively [37]. The first zinc finger domain containing seven zinc finger motifs is located at the amino terminus and the second one consisting of three zinc finger motifs lies towards the carboxyl terminus. Both zinc finger domains bind to specific DNA sequences. The first one recognizes a consensus sequence of GA(C/T)AAGA(T/C)AAGATAA [38], whereas the second one binds to a consensus sequence of GAAGATGAG [39]. A highly acidic domain is also found near the carboxyl terminus of EVI1. Although acidic domains are often found in transcription factors as part of the transcriptional activation domains, no data have shown that the acidic domain of EVI1 is essential for transcriptional activation [39]. In addition, transcriptional repressor domains are also identified in EVI1, such as the proline-rich repression domain [39] (Figure 1). Both human and murine EVI1 complementary DNAs (cDNA) share more than 90% homology [40].
MDS1 was first identified in a fusion gene resulting from chromosomal rearrangements involving chromosome band 3q26. This 3;21 translocation leads to the expression of several fusion proteins including AML1/MDS1, AML1/EAP and AML1/MDS1/EVI1 [1,2,41,42]. The MDS1 locus is mapped 150–300 kb upstream of EVI1 at chromosome band 3q26 [2].
Fears et al. [1] performed Northern blot in multiple tissues with an MDS1 probe and showed two large bands of 5.8 and 6.2 kb, which were identical to those observed with an EVI1 probe, suggesting that MDS1 and EVI1 could be expressed as one transcript, designated as MDS1/EVI1, in normal tissues. The existence of the MDS1/EVI1 transcript was confirmed by isolating and sequencing cDNA clones from normal human pancreas and kidney libraries. Sequence analysis of the clones showed an open reading frame in which MDS1 was spliced in frame to the second exon of EVI1, resulting in a new gene, MDS1/EVI1 [1]. In EVI1, the first methionine is located in the third exon. In MDS1/EVI1, the splicing site is at the junction between exon 1 and exon 2 of EVI1 [42]. With 125 codons from MDS1 and 63 codons from the second and part of the third exon of EVI1, a total of 188 codons are added upstream of the start codon of EVI1 [42] (Figure 2). This new N-terminal extension of EVI1 has later been named the PR domain [1] (Figure 2), and shares about 40% homology with PR domains found in several proteins including C. elegans differentiation factor egl-43 [43], B cell factor positive regulatory domain I binding factor 1 (PRDI-BF1) [44,45,46] and retinoblastoma binding protein RIZ [47,48]. The homology between MDS1/EVI1 and egl-43 extends from PR domains to one of the zinc finger domains [43,49]. Experiments in C. elegans and in mice where egl-43 and MDS1/EVI1 were disrupted by homologous recombination showed that disruption of either protein prohibits the development of the nervous system [43,49], further suggesting the close relationship between these two proteins.
AML1 contains a runt homology domain (RHD) at the N-terminus which is highly homologous to the Drosophila melanogaster segmentation gene runt and a subunit of polyomavirus enhancer binding protein 2 (PEBP2 or PAE2). A proline-serine-threonine (PST) domain is located at the C-terminus of AML1 which is required for transcriptional activation [50]. AML1/MDS1/EVI1 contains an open reading frame of 4185 nucleotides and encodes a 1395 amino acid protein [3]. In AML1/MDS1/EVI1, AML1 is interrupted at the end of RHD, followed by the entire MDS1/EVI1 cDNA. Therefore, the AML1/MDS1/EVI1 fusion protein is a chimeric protein which consists of the RHD of AML1, the PR domain and two zinc finger domains of MDS1/EVI1 (Figure 2).
Besides MDS1/EVI1, EVI1 also has a few alternative splice transcripts which encode for truncated forms of EVI1, called EVI-Δ324 and EVI1-Δ105. Although some studies have revealed part of the function of EVI1 and MDS1/EVI1, little is known about the function of the splicing variants of EVI1.
1.2. Expression and Function of EVI1 and MDS1/EVI1 in Development
Perkins et al., 1991, first reported an expression pattern of EVI1 in embryonic and adult mouse tissues. They showed that EVI1 expression was high in lung, heart, urinary system and Mullerian ducts. Additionally, EVI1 was detectable in primary fetal cells, differentiating red blood cells, developing limbs and developing oocytes in the ovary [51]. The spatial and temporal expression pattern of EVI1 suggests that EVI1 plays an important role in mouse development. However, these studies by Northern blots were carried out with a probe that also recognizes MDS1/EVI1. Thus, it is unknown whether the results are specific for EVI1 or MDS1/EVI1 or both. Later on, Nucifora et al. [2] showed, by Northern blot with probes specific for MDS1/EVI1, that MDS1/EVI1 is expressed at high levels in adult kidney, pancreas and lung, but at lower levels in several other adult tissues including heart, placenta and skeletal muscle. However, MDS1/EVI1 was not detectable in thymus, spleen, leukocytes, peripheral blood or myeloid [2].
More studies carried out by Sitailo et al. [52] showed that expression of EVI1 and MDS1/EVI1 is regulated independently during embryonic stem cell differentiation. EVI1 was not detectable in undifferentiated totipotent embryonic stem cells. During embryonic stem cell differentiation, EVI1 appears on day 3 and disappears on day 13. MDS1/EVI1 is expressed at low levels in undifferentiated totipotent embryonic stem cells. Expression of MDS1/EVI1 is at the highest on day 7 of differentiation and persists for about 14 days [52].
Bartholomew et al. [4] showed for the first time that EVI1 functions as a transcription repressor. They used deletion mutagenesis to show that the repressor activity of EVI1 lies between amino acids 514 and 714 [4]. Fusion proteins lacking this region retain nuclear localization, indicating that loss of transcriptional repression activity is not due to changes in subcellular localization [4]. Soderholm et al. [5] compared the transcriptional activity of MDS1/EVI1 and EVI1 and showed that MDS1/EVI1 and EVI1 both recognize and bind to the consensus sequence of the first zinc finger domain. By using deletion mutants, they demonstrated that the binding was due to the proximal region of zinc fingers [5]. Using reporter assays in which CAT gene expression was controlled by a 207 bp genomic promoter with nine AGATA repeats, they also showed that activation of the promoter by MDS1/EVI1 was repressed by EVI1 [5]. In addition, another group demonstrated that MDS1/EVI1 increased GATA-binding factor 1 (GATA-1) transcriptional activity whereas EVI1 acted as a repressor of GATA-1 [53]. Therefore, removal of the N-terminal extension of 188 amino acids converts MDS1/EVI1 from a transcriptional activator to a repressor. Further work indicated that the PR domain, which is absent in EVI1, is responsible for the activating properties of MDS1/EVI1, because it functions as a transcriptional activation domain when fused with the DNA binding domain of Gal-4 [5]. Taken together, EVI1 and MDS1/EVI1 are two different proteins with large overlaps. While EVI1 is a transcriptional repressor, MDS1/EVI1 is a strong transcriptional activator.
EVI1 is involved in cell proliferation, vascularization and differentiation [49]. The full-length EVI1 transcript was disrupted by targeted mutagenesis in embryonic stem cells and showed that embryos with the EVI1 homozygous mutant died on day 10.5. At that time, mutant embryos were easily differentiated from wild type embryos by hemorrhaging, widespread hypocellularity and failure in the development of paraxial mesenchyme [49]. Additionally, the peripheral nervous system failed to develop in mutant embryos and defects in somites, heart and cranial ganglia were detected [49]. The defects in cellular proliferation observed in mutant embryos indicate that EVI1 is involved in the regulation of cell growth. A wide range of defects agree with the expression pattern of EVI1 during embryonic development [51]. However, at the time of the study, the existence of MDS1/EVI1 was unknown. Therefore, this study only demonstrates the importance of the gene locus shared by EVI1 and MDS1/EVI1 during embryonic development.
In an attempt to differentiate EVI1 and MDS1/EVI1 function, Zhang et al. [54] generated a mouse model in which MDS1/EVI1 expression was eliminated while expression of EVI1 remained unchanged. This study and others demonstrate that EVI1 and MDS1/EVI1 may exert their functions in modulating hematopoiesis at different stages and by different mechanisms [9,54].
Taken together, these results suggest that MDS1/EVI1 is expressed in normal tissues rather than EVI1, and that the inappropriate expression of EVI1, an altered activator which has become a repressor, could be the cause of leukemic transformation [5]. Although the expression and function of MDS1/EVI1 in malignancy remain elusive, EVI1 has been extensively studied in cancer, especially leukemia.
1.3. EVI1 in Myeloid Leukemias
EVI1 has been established as a marker in myeloid leukemias for aggressive disease with poor survival [8,10,11,12,13,55]. Chromosomal rearrangements involving 3q with EVI1 overexpression are found in 2–2.5% AML cases, whereas EVI1 overexpression is seen in 6–11% of adult AML [14,15,16,17]. Both inv(3)/t(3;3) and EVI1 overexpression alone are well-established prognostic markers in AML associated with poor outcomes [15,16,17]. In the most recent 2008 World Health Organization (WHO) classification, AML with inv(3)/t(3;3)(q21q26) has been categorized as a distinct entity characterized by its aggressive course and poor prognosis [56]. In a study of 319 de novo AML patients, high expression of EVI1 but not MDS1/EVI1 was associated with highly aggressive AML [15]. In another study of 266 AML patients, it was shown that both EVI1 and MDS1/EVI1 overexpression predict short remission duration [57]. In CML, EVI1 overexpression has been associated with the higher self-renewal capacity of CML stem cells and resistance to tyrosine kinase inhibitors in murine models [11]. The prognostic value of AML1/MDS1/EVI1 in leukemia has yet to be determined.
1.4. Chromosomal Rearrangements Activate EVI1 Transcription in Leukemia
EVI1 expression is not detectable in the bone marrow of normal individuals. However, EVI1 transcription has been found to be inappropriately activated in mouse and human hematopoietic cells of myeloid leukemia [58]. In human leukemia, activation of EVI1 transcription is usually caused by chromosomal rearrangements at the site of the EVI1 locus, 3q26 [59,60], among which, the most frequent involve 3q21 and 3q26, resulting in t(3;3)(q21q26) and inv(3)(q21q26). Using pulsed field gel electrophoresis (PFGE) and fluorescence in situ hybridization (FISH), the breakpoints of t(3;3) have been mapped at the 5′ end of EVI1, whereas the breakpoints of inv(3) have been mapped at the 3′ end [61,62]. Due to the specific orientation of the two breakpoints on chromosome 3, it was previously speculated that the activation may result from juxtaposition of EVI1 to the enhancer of Ribophorin I, a constitutively expressed housekeeping gene, to the coding region of EVI1 [62]. Later, it was shown that translocation of a GATA2 enhancer to 3q26 results in overexpression of EVI1 [63,64].
Activation of EVI1 transcription can also result from translocations involving chromosomes other than 3, leading to the constitutive expression of a fusion protein in which the entire EVI1 locates at the carboxyl end. The most frequent cases are t(3;21)(q26;q22) and t(3;12)(q26;p13) [2,3,65], which are seen in myelodysplastic syndromes (MDS), AML or CML during the blast crises [66,67,68,69]. t(3;21) results in fusion between the DNA binding domain of AML1 and MDS1/EVI1 [2,3], whereas t(3;12) leads to the fusion between the amino terminus of the ETS protein TEL to MDS1/EVI1 [70,71,72]. While AML1 and TEL are related to a large number of frequent chromosomal rearrangements in myeloid and lymphoid leukemias [73,74,75], EVI1 has been associated with chromosomal rearrangements in myeloid leukemias exclusively. Both AML1/MDS1/EVI1 and TEL/MDS1/EVI1 are transcribed by the promoter of AML1 or TEL.
2. EVI1, MDS1/EVI1 and AML1/MDS1/EVI1 in Leukemogenesis
2.1. EVI1 and MDS1/EVI1 in Leukemogenesis
2.1.1. Differentiation
Overexpression of EVI1 prevented 32Dcl3 cells from expressing myeloperoxidase and differentiating to granulocytes after granulocyte colony stimulating factor (G-CSF) treatment [52]. Later Khanna-Gupta et al. [76] showed that 32Dcl3 cells contain a rearrangement at the EVI1 locus and constitutively overexpress EVI1. EVI1 expression decreased only slightly during G-CSF-induced myeloid maturation. Activation of EVI1 transcription in naive 32Dcl3 cells possibly contributes to the immortalization of the cell line by impairing spontaneous differentiation. These results do not conflict with the observation from Sitailo et al. [52] that overexpression of EVI1 blocks differentiation in 32Dcl3 cells in response to G-CSF, but rather suggest that higher levels of EVI1 are required to block G-CSF-induced differentiation than spontaneous differentiation in myeloid cells [52].
Overexpression of EVI1 in primary hematopoietic cells from mouse bone marrow decreased their ability to form burst-forming units-erythroid (BFU-E) in semisolid medium [53]. Thus, a potential mechanism for the leukemogenic and myelodysplastic effects of EVI1 is the inhibition of the differentiation of hematopoietic cells. Louz et al. [77] showed that disruption of erythropoiesis is seen in one of three mouse lines of transgenic mice overexpressing EVI1.
Glass et al. [78] conducted a comprehensive genome-wide study of EVI1 DNA binding sites in leukemic cells and compared whole transcriptome gene expression profiles between EVI1-overexpressed and EVI1 knockdown leukemic cells using chromatin immunoprecipitation-Sequencing (ChIP-Seq) and RNA-Sequencing (RNA-Seq) expression profiling. They showed that EVI1 can directly bind to and downregulate the master myeloid differentiation gene, Cebpe. Previous studies have shown that Cebpe plays a critical role in the terminal differentiation of granulocytes [79,80,81]. In two different EVI1-overexpressing leukemic cell lines, DA-1 cells and NFS-60 cells, Cebpe was found to be downregulated by 2-fold. Several Cebpe downstream target genes, including Epx, Lcn2, Mmp8 and Prg2, were also significantly downregulated in both EVI1 leukemic cells [78].
2.1.2. Apoptosis
Besides terminal myeloid differentiation, Glass et al. [78] showed that EVI1 target genes are also related to apoptosis. They identified seven significantly downregulated genes that encode for ligand gated p2 purinoreceptors, including P2rx3, Prx4, and P2rx7. They showed that EVI1 binds to three different sites within the P2rx7 promoter region and significantly downregulates P2rx7 transcription in AML [78]. P2RX7 is a cell surface ATP receptor mainly expressed in macrophages and neutrophils and mediates ATP-induced apoptosis of macrophages and neutrophils. It has been shown that loss of function of the P2RX7 receptor impaired apoptosis [82,83,84,85]. Activation of the P2RX7 receptor leads to activation of caspase-1 [86]. Humphreys et al. [87] showed that P2RX7 stimulation in response to ATP rapidly increases caspase-3 protease activity, which is associated with DNA fragmentation and upregulation of the c-Jun N-terminal kinase pathway [87].
Perkins et al. [88] investigated the physiological effects of EVI1 knockdown in murine leukemic cells expressing EVI1. They showed that knockdown of EVI1 induces apoptosis via the intrinsic pathway but not the extrinsic pathway. Specifically, procaspase 3 and 9 were cleaved but caspase 8 or Bid remained unchanged. DNA fragmentation and histone release were both induced, and mitochondrial membrane potential was reduced.
2.1.3. Cell Quiescence
Zhang et al. [54] generated an MDS1/EVI1 knockout mouse model in which hematopoietic stem cells (HSC) had a high proliferation rate, indicating HSCs are shifted from quiescence to active cycling, leading to a reduction in the number of HSCs and a loss of long-term repopulation capacity. RNA sequencing analysis in HSCs of MDS1/EVI1-KO mice revealed a significant decrease in the expression of Cdkn1c, an important negative growth regulator that encodes for p57-Kip2, which preferentially inhibits cyclin E-Cdk2, a G1 cyclin complex. Reintroduction of MDS1/EVI1 into the HSCs normalizes both low-expression p57-Kip2 and the high level of cell proliferation [54]. However, the mechanism by which MDS1/EVI1 regulates Cdkn1c transcription has not been elucidated.
To further investigate the effects of EVI1 overexpression in HSCs, Glass et al. [89] generated a mouse model in which EVI1 can be induced in the HSC compartment. In this model, supplementation of doxycycline can lead to an upregulation of EVI1 transcripts of 10,000-fold, whereas MDS1/EVI1 transcripts remain unchanged. The upregulation of EVI1 caused cell cycle arrest in HSCs [89]. Kustikova et al. [90] used a Rosa26rtTA-nls-Neo2 mouse model and overexpressed EVI1 in hematopoietic progenitor cells. They showed that inducible expression of EVI1 leads to cell cycle arrest in G0/G1 in hematopoietic progenitor cells. Gene expression microarray showed enhanced expression of cell cycle inhibitory genes Cdkn1b and Cdkn1c and downregulation of cyclins and their kinases [90]. These data suggest that EVI1 may contribute to the enlargement of a population of quiescent hematopoietic stem cells.
Studies by Yamakawa et al. [91] showed that overexpression of EVI1 inhibits cell growth and causes cells to accumulate in the G0 phase in. Konrad et al. [92] showed that induction of either EVI1 or MDS1/EVI1 inhibits cell proliferation by slowing the transit through the cell mitotic cycle. Induction of either protein drives cells to accumulate at the G0/G1 phase and moderately increased the rate of spontaneous apoptosis. Overexpression of EVI1 in U20S cells induced the accumulation of supernumerary centrosomes in cells and resulted in a cell cycle arrest in the G0/G1 phase. In addition, EVI1-overexpressing cells showed higher levels of cyclin D1 and p21, reduced Cdk2 activity and activated p53 pathway [93,94]. These data suggest that EVI1 may play a role in cell quiescence.
2.2. AML1/MDS1/EVI1 in Leukemogenesis
2.2.1. Repression of TGF-β-Mediated Growth Inhibition
AML1/MDS1/EVI1 inhibits transactivation of TGF-β-responsive promoters similar to EVI1. Expression of EVI1 or AML1/MDS1/EVI1 in 32Dcl3 cells overcomes TGF-β-mediated cell growth inhibition. AML1/MDS1/EVI1 can physically bind to SMAD3 and inhibit its activity to induce transcription of TGF-β target genes. AML1/MDS1/EVI1 also interacts with CtBP through the CtBP-binding consensus sequence PLDLS, which is involved in TGF-β-mediated gene transcription [7].
2.2.2. Stimulation of Proliferation
Kurokawa et al. [95] introduced AML1/MDS1/EVI1 into Rat1 fibroblasts and showed that AML1/MDS1/EVI1 expressing Rat1 cells form macroscopic colonies in soft agar, whereas control cells produce tiny, barely macroscopic colonies, suggesting that AML1/MDS1/EVI1 is a transforming gene. They also demonstrated that introduction of AML1/MDS1/EVI1 into Rat1 clones harboring BCR/ABL conferred a higher capacity for anchorage-independent growth. Deletion mutant analysis showed that removal of the second zinc finger domain of EVI1 completely abrogated the ability of AML1/MDS1/EVI1 to transform Rat1 cells [95]. The transforming effect was due to AP-1 activation by AML1/MDS1/EVI1 [96]. These results suggest that AML1/MDS1/EVI1 could play a pivotal role in the development of chronic myelogenous leukemia [6].
3. EVI1 and MDS1/EVI1 in Solid Tumors
Besides leukemia, EVI1 has been linked to other cancers as well. In recent years, it has been shown that EVI1 is also involved in the occurrence and progression of some solid tumors, including glioblastoma, squamous cell lung cancer, ovarian cancer, prostate cancer and breast cancer. Hou et al. [97] assessed the clinical significance of the MDS1 and EVI1 complex locus protein (MECOM) in glioblastoma multiforme (GBM). They showed that MECOM is highly expressed in 41.9% of GBM tumor samples using immunohistochemistry. In addition, MECOM mRNA expression is also higher in tumor tissues than in normal tissues. They further indicated that higher expression of MECOM was associated with a lower overall survival rate. The one-year survival rate of the MECOM high expression group was three times lower than that in the MECOM low expression group. In addition, they identified MECOM expression as an independent prognosis marker in GBM [97]. Xu et al. [18] showed that 32.32% of squamous cell lung cancer samples express a high level of EVI1. High expression of EVI1 was significantly associated with a poorer five-year survival rate of squamous cell lung cancer patients. Moreover, EVI1 was identified as an independent prognostic factor, suggesting that EVI1 alone was enough to predict poor prognosis of squamous cell lung cancer [18]. Another study done by Wang et al. [19] demonstrated EVI1 overexpression in both estrogen receptor-positive (ER +) and estrogen receptor-negative (ER −) breast carcinomas by analyzing a tissue microarray of 608 breast carcinoma patient specimens. Prognostic relevance of EVI1 overexpression was shown in triple-negative breast carcinoma but not in the HER2-positive breast carcinoma subset [19]. In head and neck squamous cell carcinomas, higher EVI1 expression is associated with a higher rate of lymph node metastasis [98].
Redistribution of EVI1 has also been reported in different types of cancer. Using immunohistochemistry, Brooks et al. [25] showed a relative redistribution of EVI1 from the cytoplasm of normal oocytes to increased nuclear and diffused cytoplasmic localization in ovarian tumors. Higher levels of EVI1 mRNA were also detected in ovarian carcinoma compared to the normal ovary, suggesting EVI1 could play a role in ovarian cancer initiation and progression. Similarly, EVI1 also exhibits a redistribution of expression in prostate cancer. EVI1 is expressed in the stem cell compartment which is located at the basal layer. However, EVI1 was distributed heterogeneously within samples in a prostate cancer progression cohort consisting of 219 samples from patients with primary prostate cancer, lymph node and distant metastases. EVI1 expression is associated with tumor progression, suggesting EVI1 expression may be a driver event in prostate cancer [26].
Functionally, knockdown of EVI1 enhanced sensitivity to apoptosis, and inhibited cell cycle progression, proliferation, migration and anchorage-independent growth in human prostate cancer cells. Interestingly, they also showed that EVI1 is involved in the regulation of stem cell properties. While EVI1 expression was upregulated in experimentally derived docetaxel-resistant prostate cancer cells, knockdown of EVI1 restored the sensitivity to docetaxel in these cells. These data suggest that EVI1 may regulate prostate cancer progression and therapy resistance through mediating stem cell properties [26]. In human breast cancer cells, knockdown of EVI1 inhibited proliferation, tumorigenicity and apoptosis resistance. These effects were rescued by estrogen addition in ER + breast carcinoma cells. In addition, estrogen supplementation restored phosphorylated extracellular signal-regulated kinases (pERK) expression in EVI1 knockdown cells, indicating that EVI1 and estradiol signaling merge on MAPK activation. On the contrary, knockdown of EVI1 had no effect on constitutive ERK activity in HER2-positive breast carcinoma cells. By analyzing the transcriptome of control and EVI1 knockdown MDA-MB-231 cells using gene expression microarrays, G-protein-coupled receptor signaling pathways were identified as the most influenced pathways. Investigation of the direct target genes of EVI1 revealed that GPR54-ligand KISS1 was a strong candidate among others. ChIP assays revealed that the KISS1 promoter is a novel target for EVI1 in breast carcinoma. Thus, EVI1 directly modulates G protein-coupled receptors (GPCRs) signaling by targeting the GPR54 ligand KISS1 at the transcriptional level. In addition, pathways involved in cell cycle control and progression, apoptosis resistance and receptor tyrosine-protein kinase ErbB-2 signaling were also affected by EVI1 [19].
Deng et al. [20] assessed the EVI1 expression level in 15 pairs of human colorectal cancer (CRC) samples and 3 pairs of colon adenocarcinoma samples and their adjacent normal mucosal controls. They showed that EVI1 was overexpressed in 53% of CRC samples (8/15) and 100% of colon adenocarcinoma samples. In addition, they determined the EVI1 protein level in human colon cancer cell lines and revealed that EVI1 was overexpressed in 100% of the CRC cell lines tested (5/5), including Caco2, DLD1, HCT116, HT29 and Lovo. EVI1 represses TGF-β signaling and antagonizes its growth inhibitory effect in colon cancer cells [20]. Liu et al. [21] reported that EVI1 is overexpressed in some colon cancer cell lines including HT29 cells and Caco2 cells. Knockdown of EVI1 by small interfering RNA (siRNA) decreased AKT phosphorylation in HT29 cells and increased their sensitivity to taxol-mediated apoptosis [21]. EVI1 directly binds to the B-cell lymphoma-extra large (Bcl-xL) promoter element via the first zinc finger domain and thus inhibits apoptosis. ChIP assays revealed that EVI1 interacts with the Bcl-xL promoter. Knockdown of EVI1 downregulated Bcl-xL expression. In addition, co-expression of EVI1 with P300/CBP-associated factor (PCAF) abolishes the effect of EVI1 on Bcl-xL, whereas co-expression of EVI1 and the dominant-negative form of PCAF showed no effect on EVI1 activity, suggesting that acetylation of EVI1 abrogated its ability to bind to the Bcl-xL promoter and promote Bcl-xL expression [22]. EVI1 delays cell proliferation and cell cycle progression. Compared to control siRNA, knockdown of EVI1 with EVI1-specific siRNA decreased the number of cells in the G0/G1 phase and increased the number of cells in the S and G2/M phases by 12–14%. They revealed that EVI1 directly binds to the DeltaNp63 promoter element and downregulates its expression. Downregulation of DeltaNp63 promotes p21 expression in HT29 cells and HCT116 cells, as well as in colon cancer patient samples with a low level of p53 [23]. Nayak et al. [24] later analyzed colon cancer patient microarray datasets and observed a negative correlation between EVI1 expression level and several epithelial-mesenchymal transition (EMT)-related markers, including SLUG, ZEB1, ZEB2, SNAIL, TWIST1 and TWIST2. Later, they performed a ChIP assay and luciferase reporter assays and showed that EVI1 directly binds to the SLUG promoter element via the second zinc finger domain and downregulates its expression. Invasion assays revealed that knockdown of EVI1 increased invasion activity and resulted in EMT-like morphological features such as a spindle-shaped appearance with a significant downregulation of the epithelial marker E-CADHERIN and upregulation of the mesenchymal marker N-CADHERIN in COLO205 cells. Injection of these EMT-induced COLO205 cells into mice failed to show metastasis in any organ, whereas injection of the EVI1-positive non-EMT COLO205 cells showed metastasis in the intraperitoneal layer as well as in the lungs and spleen. These studies demonstrate that EVI1 inhibits EMT by suppressing SLUG transcription. Inhibition of EMT does not abolish the ability of EVI1 to promote tumorigenicity and metastasis in colon cancer [24].
In summary, EVI1 and its variants play important functions in leukemia and solid tumors, as summarized in Figure 3.
4. Downstream Signaling Pathways
Although the molecular mechanism by which EVI1 promotes leukemia transformation is still unclear, many studies have been conducted in different cell systems to investigate the downstream signaling pathways of EVI1, by which it exerts its biological functions (Figure 4). Studies have shown that EVI1 represses transforming growth factor β (TGF-β) signaling and antagonizes the growth-inhibitory effects of TGF-β. [27]. The transcriptional repression of EVI1 is mediated by the interaction of EVI1 with SMAD3 via zinc fingers 1–7 [27]. Deletions of zinc fingers 1–7 of EVI1 abolish its ability to bind to SMAD3 and to inhibit TGF-β-mediated transactivation activity [27]. In addition, Izutsu et al. [28] demonstrated that TGF-β activation of the reporters is partially impaired by an EVI1 missense mutant which lacks the ability of CtBP binding, suggesting the interaction of EVI1 with CtBP through the EVI1 repression domain is also necessary for the repressive effect [28]. EVI1 has also been shown to interact with other SMADs, including SMAD2 and SMAD4, suggesting that EVI1 may play a regulatory role in signaling mediated by other TGF-β family members such as activin and BMPs [29]. Further studies are needed to determine whether that is the case.
EVI1 represses the transcription of PTEN in murine bone marrow, which results in the activation of the AKT/ mTOR signaling [30]. Knockdown of EVI1 increased the PTEN protein level and decreased phosphorylation of AKT and mTOR [30]. ChIP assays revealed that EVI1 interacts with several polycomb group proteins and recruits polycomb-repressive complexes to the PTEN promoter region, which induces histone modification to repress PTEN transcription. It represents a novel epigenetic mechanism of AKT/mTOR activation in leukemia [30]. These results indicate that the interaction between the PTEN/AKT/mTOR signaling pathway and the EVI1-polycomb complexes could be potential therapeutic targets for leukemia with activated EVI1 [30]. Knockdown of EVI1 also represses cell proliferation and promotes apoptosis via the PTEN/AKT signaling pathway in hilarcholangiocarcinoma [31].
In addition, EVI1 increases AP-1 activity by promoting transactivation of the c-fos promoter [32]. Deletion mutants of EVI1 were constructed to demonstrate that the second zinc finger domain is essential for AP-1 activation and transactivation of the c-fos promoter [32]. Later, Kurokawa et al. [33] showed that blocking of the ERK pathway by a catalytically inactive form of ERK or a dominant-negative form of MEK1 markedly repressed EVI1-induced AP-1 activity, suggesting that ERK signaling is essential for the efficient induction of AP-1 by EVI1.
EVI1 acts as an inhibitor of c-Jun N-terminal kinase (JNK), which is a class of mitogen-activated protein kinases implicated in cell stress response [33]. EVI1 physically interacts with JNK via its first zinc finger domain but does not affect JNK phosphorylation [33]. However, this interaction is required for JNK inhibition [33]. In addition, EVI1 protects cells from stress-induced cell death dependent upon its ability to inhibit JNK [33]. These results indicate that EVI1 blocks cell death by selectively inhibiting JNK, thus promoting oncogenic transformation [33].
EVI1 completely abrogates the anti-proliferative and apoptotic effects of IFN-α in hematopoietic cells [34]. EVI1 prolongs STAT1 phosphorylation and binding to the first exon of promyelocytic leukemia protein (PML), thereby repressing PML transcription and preventing the activation of PML-dependent apoptotic pathways, resulting in the loss of IFN-α response [34].
Xu et al. [35] showed that EVI1 negatively regulates the nontypeable Haemophilus influenzae- and TNF-α-induced NF-κB activation and subsequent inflammatory responses by inhibiting the DNA-binding activity of the NF-κB complex. EVI1 directly interacts with the NF-κB p65 subunit and inhibits its acetylation at lysine 310, which leads to the inhibition of its DNA-binding activity [35]. In addition, nontypeable Haemophilus influenzae and TNF-α can in turn induce the expression of EVI1 in an NF-κB-dependent manner, thereby unveiling a novel inducible negative feedback loop in NF-κB-dependent inflammation [35].
5. Conclusions
EVI1 is a 1051 amino acid transcription factor that has been extensively studied due to its association with myeloid leukemia. EVI1 contains two zinc finger domains, a proline-rich repressor domain and a C-terminus acidic domain [38,39]. Both zinc finger domains bind to specific DNA sequences [38,39]. MDS1/EVI1 is encoded by the same locus as EVI1 with the complete sequence of EVI1 and an N-terminal extension of 188 amino acids including 125 codons from MDS1 and 63 codons derived from the second exon and part of the third exon of EVI1 [99]. The extension region shares homology with the PR domain of RIZ and B-LYMP1 proteins but its function is unclear [44,45,46,47,48]. Previous studies have described EVI1 as a transcriptional repressor, whereas MDS1/EVI1 is a transcriptional activator [4,5].
EVI1 expression is not detectable in the bone marrow of normal individuals. However, in leukemia, EVI1 transcription has been found to be inappropriately activated in hematopoietic cells [58,100]. Activation of EVI1 transcription is associated with chromosomal rearrangements resulting in either overexpression of the EVI1 protein, or constitutive expression of a fusion protein, AML1/MDS1/EVI1, in which part of AML1 fuses to the N-terminus of the entire MDS1/EVI1. AML1/MDS1/EVI1 is a 1395 amino acid protein with an open reading frame of 4185 nucleotides [3]. In AML1/MDS1/EVI1, AML1 is interrupted at the end of the RHD (runt homology domain) [3], followed by the entire MDS1/EVI1 cDNA. Therefore, the AML1/MDS1/EVI1 fusion protein is a chimeric protein which consists of the RHD, PR domain and two zinc finger domains of MDS1/EVI1. AML1/MDS1/EVI1 has a similar function to EVI1 as a transcription factor [6,7]. Studies have also shown that EVI1 and AML1/MDS1/EVI1 both play an important role in leukemogenesis.
Author Contributions
Conceptualization, B.L. and J.W.; manuscript writing, B.L.; manuscript editing, J.W. All authors have read and agreed to the published version of the manuscript.
Funding
The work is supported by NIH grants R01CA212241, R01CA215389 and R01CA208063 to J.W.
Acknowledgments
We acknowledge Tanvi Brar for her help in editing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fears, S.; Mathieu, C.; Zeleznik-Le, N.; Huang, S.; Rowley, J.D.; Nucifora, G. Intergenic splicing of MDS1 and EVI1 occurs in normal tissues as well as in myeloid leukemia and produces a new member of the PR domain family. Proc. Natl. Acad. Sci. USA 1996, 93, 1642–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nucifora, G.; Begy, C.R.; Kobayashi, H.; Roulston, D.; Claxton, D.; Pedersen-Bjergaard, J.; Parganas, E.; Ihle, J.N.; Rowley, J.D. Consistent intergenic splicing and production of multiple transcripts between AML1 at 21q22 and unrelated genes at 3q26 in (3;21)(q26;q22) translocations. Proc. Natl. Acad. Sci. USA 1994, 91, 4004–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, K.; Ogawa, S.; Tanaka, T.; Miyoshi, H.; Kurokawa, M.; Mano, H.; Yazaki, Y.; Ohki, M.; Hirai, H. Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. EMBO J. 1994, 13, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, C.; Kilbey, A.; Clark, A.M.; Walker, M. The Evi-1 proto-oncogene encodes a transcriptional repressor activity associated with transformation. Oncogene 1997, 14, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderholm, J.; Kobayashi, H.; Mathieu, C.; Rowley, J.D.; Nucifora, G. The leukemia-associated gene MDS1/EVI1 is a new type of GATA-binding transactivator. Leukemia 1997, 11, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Tanaka, T.; Kurokawa, M.; Imai, Y.; Ogawa, S.; Mitani, K.; Yazaki, Y.; Hirai, H. The AML1/ETO(MTG8) and AML1/Evi-1 Leukemia-Associated Chimeric Oncoproteins Accumulate PEBP2β(CBFβ) in the Nucleus More Efficiently Than Wild-Type AML1. Blood 1998, 91, 1688–1699. [Google Scholar] [CrossRef]
- Senyuk, V.; Chakraborty, S.; Mikhail, F.M.; Zhao, R.; Chi, Y.; Nucifora, G. The leukemia-associated transcription repressor aml1/mds1/evi1 requires ctbp to induce abnormal growth and differentiation of murine hematopoietic cells. Oncogene 2002, 21, 3232–3240. [Google Scholar] [CrossRef]
- Arai, S.; Yoshimi, A.; Shimabe, M.; Ichikawa, M.; Nakagawa, M.; Imai, Y.; Goyama, S.; Kurokawa, M. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood 2011, 117, 6304–6314. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, K.; Sato, T.; Yoshimi, A.; Goyama, S.; Tsuruta, T.; Kobayashi, H.; Shimabe, M.; Arai, S.; Nakagawa, M.; Imai, Y.; et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J. Exp. Med. 2011, 208, 2403–2416. [Google Scholar] [CrossRef] [Green Version]
- Konantz, M.; André, M.C.; Ebinger, M.; Grauer, M.; Wang, H.; Grzywna, S.; Rothfuss, O.C.; Lehle, S.; Kustikova, O.S.; Salih, H.R.; et al. EVI-1 modulates leukemogenic potential and apoptosis sensitivity in human acute lymphoblastic leukemia. Leukemia 2013, 27, 56–65. [Google Scholar] [CrossRef]
- Sato, T.; Goyama, S.; Kataoka, K.; Nasu, R.; Tsuruta-Kishino, T.; Kagoya, Y.; Nukina, A.; Kumagai, K.; Kubota, N.; Nakagawa, M.; et al. Evi1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene 2014, 33, 5028–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, G.; Rommer, A.; Steinleitner, K.; Etzler, J.; Hackl, H.; Heffeter, P.; Tomasich, E.; Filipits, M.; Steinmetz, B.; Topakian, T.; et al. EVI1 promotes tumor growth via transcriptional repression of MS4A3. J. Hematol. Oncol. 2015, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wang, H.; Deng, J.; Zheng, X.; Ling, Y.; Gong, Y. Prognostic significance of the EVI1 gene expression in patients with acute myeloid leukemia: A meta-analysis. Ann. Hematol. 2019, 98, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Rogers, J.R.; Harrison, G.; Wheatley, K.; Walker, H.; Bain, B.J.; Burnett, A.K.; Goldstone, A.H.; Linch, D.C.; Grimwade, D.; et al. EVI1 expression in acute myeloid leukaemia. Br. J. Haematol. 2001, 112, 208–211. [Google Scholar] [CrossRef]
- Van Waalwijk van Doorn-Khosrovani, S.B.; Erpelinck, C.; Van Putten, W.L.J.; Valk, P.J.M.; Van der Poel-van de Luytgaarde, S.; Hack, R.; Slater, R.; Smit, E.M.E.; Beverloo, H.B.; Verhoef, G.; et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: A study of 319 de novo AML patients. Blood 2002, 101, 837–845. [Google Scholar] [CrossRef]
- Lugthart, S.; Van Drunen, E.; Van Norden, Y.; Van Hoven, A.; Erpelinck, C.A.J.; Valk, P.J.M.; Beverloo, H.B.; Löwenberg, B.; Delwel, R. High EVI1 levels predict adverse outcome in acute myeloid leukemia: Prevalence of EVI1 overexpression and chromosome 3q26 abnormalities underestimated. Blood 2008, 111, 4329–4337. [Google Scholar] [CrossRef] [Green Version]
- Gröschel, S.; Lugthart, S.; Schlenk, R.F.; Valk, P.J.M.; Eiwen, K.; Goudswaard, C.; Van Putten, W.J.L.; Kayser, S.; Verdonck, L.F.; Lübbert, M.; et al. High EVI1 Expression Predicts Outcome in Younger Adult Patients With Acute Myeloid Leukemia and Is Associated With Distinct Cytogenetic Abnormalities. J. Clin. Oncol. 2010, 28, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, S.; Ji, X. Overexpression of ecotropic viral integration site-1 is a prognostic factor of lung squamous cell cancer. OncoTargets Ther. 2017, 10, 2739–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Schäfer, T.; Konantz, M.; Braun, M.; Varga, Z.; Paczulla, A.M.; Reich, S.; Jacob, F.; Perner, S.; Moch, H.; et al. Prominent Oncogenic Roles of EVI1 in Breast Carcinoma. Cancer Res. 2017, 77, 2148–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Cao, Y.; Liu, Y.; Li, F.; Sambandam, K.; Rajaraman, S.; Perkins, A.S.; Fields, A.P.; Hellmich, M.R.; Townsend, C.M., Jr.; et al. Overexpression of Evi-1 oncoprotein represses TGF-β signaling in colorectal cancer. Mol. Carcinog. 2011, 52, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, L.; Ko, T.C.; Fields, A.P.; Thompson, K.J. Evi1 is a survival factor which conveys resistance to both TGFβ- and taxol-mediated cell death via PI3K/AKT. Oncogene 2006, 25, 3565–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, A.K.; Das Mohapatra, A.; Nayak, K.B.; Chakraborty, S. Acetylation of the Proto-Oncogene EVI1 Abrogates Bcl-xL Promoter Binding and Induces Apoptosis. PLoS ONE 2011, 6, e25370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, K.B.; Kuila, N.; Das Mohapatra, A.; Panda, A.K.; Chakraborty, S. EVI1 targets ΔNp63 and upregulates the cyclin dependent kinase inhibitor p21 independent of p53 to delay cell cycle progression and cell proliferation in colon cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Nayak, K.B.; Sajitha, I.S.; Kumar, T.R.S.; Chakraborty, S. Ecotropic viral integration site 1 promotes metastasis independent of epithelial mesenchymal transition in colon cancer cells. Cell Death Dis. 2018, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.; Woodward, S.; Thompson, F.; Dos Santos, B.; Russell, M.; Yang, J.-M.; Guan, X.-Y.; Trent, J.; Alberts, D.; Taetle, R. Expression of the zinc finger gene EVI-1 in ovarian and other cancers. Br. J. Cancer 1996, 74, 1518–1525. [Google Scholar] [CrossRef]
- Queisser, A.; Hagedorn, S.; Wang, H.; Schäfer, T.; Konantz, M.; Alavi, S.; Deng, M.; Vogel, W.; Von Mässenhausen, A.; Kristiansen, G.; et al. Ecotropic viral integration site 1, a novel oncogene in prostate cancer. Oncogene 2016, 36, 1573–1584. [Google Scholar] [CrossRef]
- Kurokawa, M.; Mitani, K.; Irie, K.; Matsuyama, T.; Takahashi, T.; Chiba, S.; Yazaki, Y.; Matsumoto, K.; Hirai, H. The oncoprotein Evi-1 represses TGF-β signalling by inhibiting Smad3. Nature 1998, 394, 92–96. [Google Scholar] [CrossRef]
- Izutsu, K.; Kurokawa, M.; Imai, Y.; Maki, K.; Mitani, K.; Hirai, H. The corepressor CtBP interacts with Evi-1 to repress transforming growth factor β signaling. Blood 2001, 97, 2815–2822. [Google Scholar] [CrossRef] [Green Version]
- Alliston, T.; Ko, T.C.; Cao, Y.; Liang, Y.-Y.; Feng, X.-H.; Chang, C.; Derynck, R. Repression of Bone Morphogenetic Protein and Activin-inducible Transcription by Evi-1. J. Biol. Chem. 2005, 280, 24227–24237. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Goyama, S.; Watanabe-Okochi, N.; Yoshiki, Y.; Nannya, Y.; Nitta, E.; Arai, S.; Sato, T.; Shimabe, M.; Nakagawa, M.; et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 2011, 117, 3617–3628. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-M.; Liu, Z.-L.; Qiu, B.; Xu, Y.-F.; Pan, C.; Zhang, Z.-L. Downregulation of EVI1 Expression Inhibits Cell Proliferation and Induces Apoptosis in Hilar Cholangiocarcinoma via the PTEN/AKT Signalling Pathway. J. Cancer 2020, 11, 1412–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Nishida, J.; Mitani, K.; Ogawa, S.; Yazaki, Y.; Hirai, H. Evi-1 raises AP-1 activity and stimulates c-fos promoter transactivation with dependence on the second zinc finger domain. J. Biol. Chem. 1994, 269, 24020–24026. [Google Scholar] [PubMed]
- Kurokawa, M.; Mitani, K.; Yamagata, T.; Takahashi, T.; Izutsu, K.; Ogawa, S.; Moriguchi, T.; Nishida, E.; Yazaki, Y.; Hirai, H. The Evi-1 oncoprotein inhibits c-Jun N-terminal kinase and prevents stress-induced cell death. EMBO J. 2000, 19, 2958–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonamici, S.; Li, D.; Mikhail, F.M.; Sassano, A.; Platanias, L.C.; Colamonici, O.; Anastasi, J.; Nucifora, G. EVI1 Abrogates Interferon-alpha Response by Selectively Blocking PML Induction. J. Biol. Chem. 2004, 280, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Woo, C.-H.; Steere, R.R.; Lee, B.C.; Huang, Y.; Wu, J.; Pang, J.; Lim, J.H.; Xu, H.; Zhang, W.; et al. EVI1 acts as an inducible negative-feedback regulator of NF-κB by inhibiting p65 acetylation. J. Immunol. 2012, 188, 6371–6380. [Google Scholar] [CrossRef] [Green Version]
- Mucenski, M.L.; Taylor, B.A.; Ihle, J.N.; Hartley, J.W.; Morse, H.C.; Jenkins, N.A.; Copeland, N.G. Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors. Mol. Cell. Biol. 1988, 8, 301–308. [Google Scholar] [CrossRef]
- Perkins, A.S.; Fishel, R.; Jenkins, N.A.; Copeland, N.G. Evi-1, a murine zinc finger proto-oncogene, encodes a sequence-specific DNA-binding protein. Mol. Cell. Biol. 1991, 11, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Delwel, R.; Funabiki, T.; Kreider, B.L.; Morishita, K.; Ihle, J.N. Four of the seven zinc fingers of the Evi-1 myeloid-transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol. Cell. Biol. 1993, 13, 4291–4300. [Google Scholar] [CrossRef] [Green Version]
- Funabiki, T.; Kreider, B.L.; Ihle, J.N. The carboxyl domain of zinc fingers of the Evi-1 myeloid transforming gene binds a consensus sequence of GAAGATGAG. Oncogene 1994, 9, 1575–1581. [Google Scholar]
- Morishita, K.; Parganas, E.; Douglass, E.C.; Ihle, J.N. Unique expression of the human Evi-1 gene in an endometrial carcinoma cell line: Sequence of cDNAs and structure of alternatively spliced transcripts. Oncogene 1990, 5, 963–971. [Google Scholar]
- Nucifora, G.; Begy, C.R.; Erickson, P.; Drabkin, H.A.; Rowley, J.D. The 3;21 translocation in myelodysplasia results in a fusion transcript between the AML1 gene and the gene for EAP, a highly conserved protein associated with the Epstein-Barr virus small RNA EBER 1. Proc. Natl. Acad. Sci. USA 1993, 90, 7784–7788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchi, N.; Nisson, P.E.; Watkins, P.C.; Faustinella, F.; Wijsman, J.; Hagemeijer, A. AMLI fusion transcripts in t(3;21) positive leukemia: Evidence of molecular heterogeneity and usage of splicing sites frequently involved in the generation of normalAMLI transcripts. Genes Chromosom. Cancer 1994, 11, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Garriga, G.; Guenther, C.; Horvitz, H.R. Migrations of the Caenorhabditis elegans HSNs are regulated by egl-43, a gene encoding two zinc finger proteins. Genes Dev. 1993, 7, 2097–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S. Blimp-1 is the murine homolog of the human transcriptional repressor PRDI-BF1. Cell 1994, 78, 9. [Google Scholar] [CrossRef]
- Lin, Y.; Wong, K.-K.; Calame, K. Repression of c-myc Transcription by Blimp-1, an Inducer of Terminal B Cell Differentiation. Science 1997, 276, 596–599. [Google Scholar] [CrossRef]
- Turner, C.; Mack, D.H.; Davis, M.M. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 1994, 77, 297–306. [Google Scholar] [CrossRef]
- Buyse, I.M.; Shao, G.; Huang, S. The retinoblastoma protein binds to RIZ, a zinc-finger protein that shares an epitope with the adenovirus E1A protein. Proc. Natl. Acad. Sci. USA 1995, 92, 4467–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Shao, G.; Steele-Perkins, G.; Huang, S. The Retinoblastoma Interacting Zinc Finger GeneRIZProduces a PR Domain-lacking Product through an Internal Promoter. J. Biol. Chem. 1997, 272, 2984–2991. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, P.R.; Bartholomew, C.; Davis, A.J.; Yutzey, K.E.; Gamer, L.W.; Potter, S.; Ihle, J.N.; Mucenski, M.L. The Evil proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech. Dev. 1997, 65, 55–70. [Google Scholar] [CrossRef]
- Kitabayashi, I.; Yokoyama, A.; Shimizu, K.; Ohki, M. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J. 1998, 17, 2994–3004. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.S.; Mercer, J.A.; Jenkins, N.A.; Copeland, N.G. Patterns of Evi-1 expression in embryonic and adult tissues suggest that Evi-1 plays an important regulatory role in mouse development. Development 1991, 111, 479–487. [Google Scholar] [PubMed]
- Sitailo, S.; Sood, R.; Barton, K.; Nucifora, G. Forced expression of the leukemia-associated gene EVI1 in ES cells: A model for myeloid leukemia with 3q26 rearrangements. Leukemia 1999, 13, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreider, B.L.; Orkin, S.H.; Ihle, J.N. Loss of erythropoietin responsiveness in erythroid progenitors due to expression of the Evi-1 myeloid-transforming gene. Proc. Natl. Acad. Sci. USA 1993, 90, 6454–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Stehling-Sun, S.; Lezon-Geyda, K.; Juneja, S.C.; Coillard, L.; Chatterjee, G.; Wuertzer, C.A.; Camargo, F.; Perkins, A.S. PR-domain–containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 2011, 118, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Kurokawa, M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci. 2012, 103, 1371–1377. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Haas, K.; Kundi, M.; Sperr, W.R.; Esterbauer, H.; Ludwig, W.-D.; Ratei, R.; Koller, E.; Gruener, H.; Sauerland, C.; Fonatsch, C.; et al. Expression and prognostic significance of different mRNA 5′-end variants of the oncogene EVI1 in 266 patients with de novo AML: EVI1 and MDS1/EVI1 overexpression both predict short remission duration. Genes Chromosom. Cancer 2008, 47, 288–298. [Google Scholar] [CrossRef]
- Fontenay-Roupie, M.; Bouscary, D.; Melle, J.; Viguie, F.; Picard, F.; Guesnu, M.; Dreyfus, F. Expression of the transcription factor Evi-1 in human erythroleukemia cell lines and in leukemias. Hematol. Cell Ther. 1997, 39, 5–10. [Google Scholar] [CrossRef]
- Fichelson, S.; Dreyfus, F.; Berger, R.; Melle, J.; Bastard, C.; Miclea, J.M.; Gisselbrecht, S. Evi-1 expression in leukemic patients with rearrangements of the 3q25-q28 chromosomal region. Leukemia 1992, 6, 93–99. [Google Scholar]
- Jenkins, R.B.; Tefferi, A.; Solberg, L.A.; Dewald, G.W. Acute leukemia with abnormal thrombopoiesis and inversions of chromosome 3. Cancer Genet. Cytogenet. 1989, 39, 167–179. [Google Scholar] [CrossRef]
- Morishita, K.; Parganas, E.; William, C.L.; Whittaker, M.H.; Drabkin, H.; Oval, J.; Taetle, R.; Valentine, M.B.; Ihle, J.N. Activation of EVI1 gene expression in human acute myelogenous leukemias by translocations spanning 300-400 kilobases on chromosome band 3q26. Proc. Natl. Acad. Sci. USA 1992, 89, 3937–3941. [Google Scholar] [CrossRef] [Green Version]
- Suzukawa, K.; Parganas, E.; Gajjar, A.; Abe, T.; Takahashi, S.; Tani, K.; Asano, S.; Asou, H.; Kamada, N.; Yokota, J.; et al. Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood 1994, 84, 2681–2688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; De Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; Van Der Velden, V.H.J.; Havermans, M.; Avellino, R.; Van Lom, K.; et al. A Single Oncogenic Enhancer Rearrangement Causes Concomitant EVI1 and GATA2 Deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottema, S.; Mulet-Lazaro, R.; Beverloo, H.B.; Erpelinck, C.A.; Van Herk, S.; Van Der Helm, R.; Havermans, M.; Grob, T.; Valk, P.; Bindels, E.; et al. Atypical 3q26/MECOM rearrangements genocopy inv(3)/t(3;3) in acute myeloid leukemia. Blood 2020, 136, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Raynaud, S.; Baens, M.; Grosgeorge, J.; Rodgers, K.; Reid, C.; Dainton, M.; Dyer, M.; Fuzibet, J.; Gratecos, N.; Taillan, B.; et al. Fluorescence in situ hybridization analysis of t(3; 12)(q26; p13): A recurring chromosomal abnormality involving the TEL gene (ETV6) in myelodysplastic syndromes. Blood 1996, 88, 682–689. [Google Scholar] [CrossRef] [Green Version]
- Rubin, C.; Larson, R.; Bitter, M.; Carrino, J.; Le Beau, M.; Diaz, M.; Rowley, J. Association of a chromosomal 3;21 translocation with the blast phase of chronic myelogenous leukemia. Blood 1987, 70, 1338–1342. [Google Scholar] [CrossRef] [Green Version]
- Rubin, C.; Larson, R.; Anastasi, J.; Winter, J.; Thangavelu, M.; Vardiman, J.; Rowley, J.; Le Beau, M. t(3;21)(q26;q22): A recurring chromosomal abnormality in therapy- related myelodysplastic syndrome and acute myeloid leukemia. Blood 1990, 76, 2594–2598. [Google Scholar] [CrossRef] [Green Version]
- Schneider, N.R.; Bowman, W.P.; Frenkel, E.P. Translocation (3;21)(q26;q22) in secondary leukemia. Report of two cases and literature review. Ann. Génét. 1991, 34, 256–263. [Google Scholar]
- Coyle, T.; Najfeld, V. Translocation (3;21) in Philadelphia chromosome—Positive chronic myelogenous leukemia prior to the onset of blast crisis. Am. J. Hematol. 1988, 27, 56–59. [Google Scholar] [CrossRef]
- Poirel, H.; Oury, C.; Carron, C.; Duprez, E.; Laâbi, Y.; Tsapis, A.; Romana, S.P.; Mauchauffe, M.; Le Coniat, M.; Berger, R.; et al. The TEL gene products: Nuclear phosphoproteins with DNA binding properties. Oncogene 1997, 14, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Jousset, C.; Carron, C.; Boureux, A.; Quang, C.T.; Oury, C.; Dusanter-Fourt, I.; Charon, M.; Levin, J.; Bernard, O.A.; Ghysdael, J. A domain of TEL conserved in a subset of ETS proteins defines a specific oligomerization interface essential to the mitogenic properties of the TEL-PDGFR beta oncoprotein. EMBO J. 1997, 16, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, P.; Wlodarska, I.; Baens, M.; Criel, A.; Selleslag, D.; Hagemeijer, A.; Van den Berghe, H.; Marynen, P. Fusion of ETV6 to MDS1/EVI1 as a result of t(3;12)(q26;p13) in myeloproliferative disorders. Cancer Res. 1997, 57, 564–569. [Google Scholar] [PubMed]
- Buijs, A.; Sherr, S.; Van Baal, S.; Van Bezouw, S.; Van Der Plas, D.; Van Kessel, A.G.; Riegman, P.; Deprez, R.L.; Zwarthoff, E.; Hagemeijer, A.; et al. Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene 1995, 10, 1511–1519. [Google Scholar] [PubMed]
- Romana, S.P.; Mauchauffe, M.; Le Coniat, M.; Chumakov, I.; Le Paslier, D.; Berger, R.; Bernard, O.A. The t(12;21) of acute lymphoblastic leukemia results in a tel-AML1 gene fusion. Blood 1995, 85, 3662–3670. [Google Scholar] [CrossRef] [Green Version]
- Golub, T.R.; Barker, G.F.; Lovett, M.; Gilliland, D. Fusion of PDGF receptor β to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell 1994, 77, 307–316. [Google Scholar] [CrossRef]
- Khanna-Gupta, A.; Lopingco, M.C.; Savinelli, T.; Zibello, T.; Berliner, N.; Perkins, A.S. Retroviral insertional activation of the EVI1 oncogene does not prevent G-CSF-induced maturation of the murine pluripotent myeloid cell line 32Dcl3. Oncogene 1996, 12, 563–569. [Google Scholar]
- Louz, D.; Van Den Broek, M.; Verbakel, S.; Vankan, Y.; Van Lom, K.; Joosten, M.; Meijer, D.; Löwenberg, B.; Delwel, R. Erythroid defects and increased retrovirally-induced tumor formation in Evi1 transgenic mice. Leukemia 2000, 14, 1876–1884. [Google Scholar] [CrossRef] [Green Version]
- Glass, C.; Wuertzer, C.; Cui, X.; Bi, Y.; Davuluri, R.; Xiao, Y.-Y.; Wilson, M.; Owens, K.; Zhang, Y.; Perkins, A.S. Global Identification of EVI1 Target Genes in Acute Myeloid Leukemia. PLoS ONE 2013, 8, e67134. [Google Scholar] [CrossRef] [Green Version]
- Akagi, T.; Thoennissen, N.H.; George, A.; Crooks, G.; Song, J.H.; Okamoto, R.; Nowak, D.; Gombart, A.F.; Phillip Koeffler, H. In Vivo Deficiency of Both C/EBPβ and C/EBPε Results in Highly Defective Myeloid Differentiation and Lack of Cytokine Response. PLoS ONE 2010, 5, e15419. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Watanabe, N.; Shibata, F.; Kitamura, T.; Ikeda, Y.; Handa, M. N-terminal Region of CCAAT/Enhancer-binding Protein epsilon Is Critical for Cell Cycle Arrest, Apoptosis, and Functional Maturation during Myeloid Differentiation. J. Biol. Chem. 2006, 281, 14494–14502. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, R.; Kim, G.-D.; Radomska, H.S.; Lekstrom-Himes, J.; Smith, L.T.; Antonson, P.; Tenen, D.G.; Xanthopoulos, K.G. CCAAT/enhancer binding protein ɛ is preferentially up-regulated during granulocytic differentiation and its functional versatility is determined by alternative use of promoters and differential splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 6462–6467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Stunff, H.; Auger, R.; Kanellopoulos, J.; Raymond, M.-N. The Pro-451 to Leu Polymorphism within the C-terminal Tail of P2X7 Receptor Impairs Cell Death but Not Phospholipase D Activation in Murine Thymocytes. J. Biol. Chem. 2004, 279, 16918–16926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.J.; Zhang, W.; Worthington, R.A.; Sluyter, R.; Dao-Ung, P.; Petrou, S.; Barden, J.A.; Wiley, J.S. A Glu-496 to Ala Polymorphism Leads to Loss of Function of the Human P2X7 Receptor. J. Biol. Chem. 2001, 276, 11135–11142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placido, R.; Auricchio, G.; Falzoni, S.; Battistini, L.; Colizzi, V.; Brunetti, E.; Di Virgilio, F.; Mancino, R. P2X7 purinergic receptors and extracellular ATP mediate apoptosis of human monocytes/macrophages infected with Mycobacterium tuberculosis reducing the intracellular bacterial viability. Cell. Immunol. 2006, 244, 10–18. [Google Scholar] [CrossRef]
- Suh, B.-C.; Kim, J.-S.; Namgung, U.; Ha, H.; Kim, K.-T. P2X7 nucleotide receptor mediation of membrane pore formation and superoxide generation in human promyelocytes and neutrophils. J. Immunol. 2001, 166, 6754–6763. [Google Scholar] [CrossRef] [Green Version]
- Kahlenberg, J.M.; Lundberg, K.C.; Kertesy, S.B.; Qu, Y.; Dubyak, G.R. Potentiation of Caspase-1 Activation by the P2X7 Receptor Is Dependent on TLR Signals and Requires NF-κB-Driven Protein Synthesis. J. Immunol. 2005, 175, 7611–7622. [Google Scholar] [CrossRef] [Green Version]
- Humphreys, B.D.; Rice, J.; Kertesy, S.B.; Dubyak, G.R. SAPK/JNK activation and apoptotic induction by the macrophage P2X7 nucleotide receptor. J. Biol. Chem. 2000, 275, 26792–26798. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.S.; Del Campo, J.J.; Xiao, Y.-Y.; Zhang, Y.; Lin, S.J.; Dudley, J.; Tuck, D.; Yatsula, B. EVI1 Blocks Apoptosis in DA-1 Myeloid Leukemia Cells Via Enhanced Transcription of the Prosurvival Gene Bcl2a1 (A1). Blood 2008, 112, 3805. [Google Scholar] [CrossRef]
- Glass, C.; Wilson, M.; González, R.; Zhang, Y.; Perkins, A.S. The role of EVI1 in myeloid malignancies. Blood Cells Mol. Dis. 2014, 53, 67–76. [Google Scholar] [CrossRef]
- Kustikova, O.S.; Schwarzer, A.; Stahlhut, M.; Brugman, M.H.; Neumann, T.; Yang, M.; Li, Z.; Schambach, A.; Heinz, N.; Gerdes, S.; et al. Activation of Evi1 inhibits cell cycle progression and differentiation of hematopoietic progenitor cells. Leukemia 2012, 27, 1127–1138. [Google Scholar] [CrossRef]
- Yamakawa, N.; Kaneda, K.; Saito, Y.; Ichihara, E.; Morishita, K. The increased expression of integrin α6 (itga6) enhances drug resistance in evi1 high leukemia. PLoS ONE 2012, 7, e30706. [Google Scholar] [CrossRef] [PubMed]
- Konrad, T.A.; Karger, A.; Hackl, H.; Schwarzinger, I.; Herbacek, I.; Wieser, R. Inducible expression of EVI1 in human myeloid cells causes phenotypes consistent with its role in myelodysplastic syndromes. J. Leukoc. Biol. 2009, 86, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Karakaya, K.; Herbst, F.; Ball, C.; Glimm, H.; Krämer, A.; Löffler, H. Overexpression of EVI1 interferes with cytokinesis and leads to accumulation of cells with supernumerary centrosomes in G0/1 phase. Cell Cycle 2012, 11, 3492–3503. [Google Scholar] [CrossRef] [PubMed]
- Wieser, R. New functions for ecotropic viral integration site 1 (EVI1), an oncogene causing aggressive malignant disease. Cell Cycle 2012, 11, 3915. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Ogawa, S.; Tanaka, T.; Mitani, K.; Yazaki, Y.; Witte, O.N.; Hirai, H. The AML1/Evi-1 fusion protein in the t(3;21) translocation exhibits transforming activity on Rat1 fibroblasts with dependence on the Evi-1 sequence. Oncogene 1995, 11, 833–840. [Google Scholar]
- Tanaka, T.; Mitani, K.; Kurokawa, M.; Ogawa, S.; Tanaka, K.; Nishida, J.; Yazaki, Y.; Shibata, Y.; Hirai, H. Dual functions of the AML1/Evi-1 chimeric protein in the mechanism of leukemogenesis in t(3;21) leukemias. Mol. Cell. Biol. 1995, 15, 2383–2392. [Google Scholar] [CrossRef] [Green Version]
- Hou, A.; Zhao, L.; Zhao, F.; Wang, W.; Niu, J.; Li, B.; Zhou, Z.; Zhu, D. Expression of MECOM is associated with unfavorable prognosis in glioblastoma multiforme. OncoTargets Ther. 2016, 9, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Idel, C.; Ribbat-Idel, J.; Kuppler, P.; Krupar, R.; Offermann, A.; Vogel, W.; Rades, D.; Kirfel, J.; Wollenberg, B.; Perner, S. EVI1 as a marker for lymph node metastasis in HNSCC. Int. J. Mol. Sci. 2020, 21, 854. [Google Scholar] [CrossRef] [Green Version]
- Nucifora, G. The EVI1 gene in myeloid leukemia. Leukemia 1997, 11, 2022–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, M.; Thompson, F.; Spier, C.; Taetle, R. Expression of the EVI1 gene in chronic myelogenous leukemia in blast crisis. Leukemia 1993, 7, 1654–1657. [Google Scholar]
Figure 1.
EVI1 protein structure. The numbered boxes indicate individual zinc fingers. The dotted box indicates the proline-rich repression domain. The oval indicates the acidic domain.
Figure 1.
EVI1 protein structure. The numbered boxes indicate individual zinc fingers. The dotted box indicates the proline-rich repression domain. The oval indicates the acidic domain.
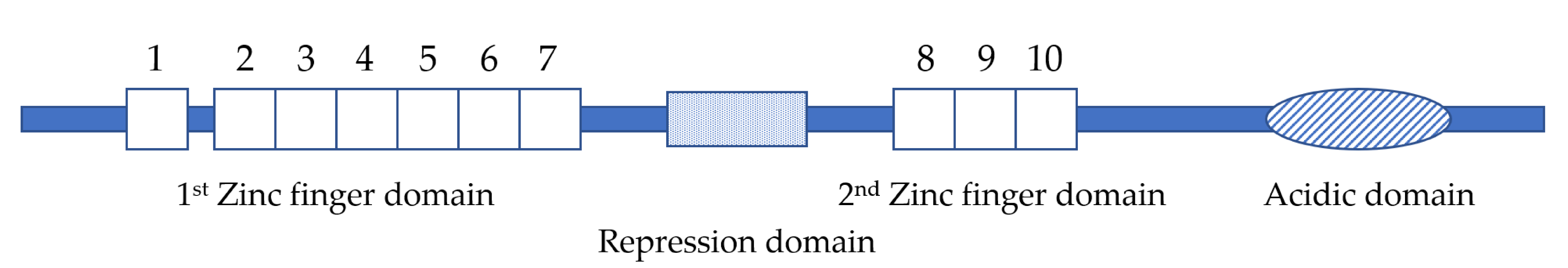
Figure 2.
Diagram illustrating the relationship between EVI1, MDS1/EVI1 and AML1/MDS1/EVI1. Untranslated regions are indicated by lines and reading frames are indicated by boxes. The vertical lines in MDS1/EVI1 indicate the junction with the 3′ alternative coding exon in MDS1, as well as the junctions between exon 1, 2 and 3 in EVI1. Numbers indicate EVI1 exons. The black thick line indicates the position of the PR domain. The vertical line in AML1/MDS1/EVI1 indicates the junction between AML1 and MDS1/EVI1, which is at the end of the runt homology domain.
Figure 2.
Diagram illustrating the relationship between EVI1, MDS1/EVI1 and AML1/MDS1/EVI1. Untranslated regions are indicated by lines and reading frames are indicated by boxes. The vertical lines in MDS1/EVI1 indicate the junction with the 3′ alternative coding exon in MDS1, as well as the junctions between exon 1, 2 and 3 in EVI1. Numbers indicate EVI1 exons. The black thick line indicates the position of the PR domain. The vertical line in AML1/MDS1/EVI1 indicates the junction between AML1 and MDS1/EVI1, which is at the end of the runt homology domain.

Figure 3.
Functions of EVI1 in leukemia and solid tumors. EVI1 regulates differentiation, apoptosis and cell quiescence in leukemia and cell proliferation, migration, apoptosis and cell cycle in solid tumors.
Figure 3.
Functions of EVI1 in leukemia and solid tumors. EVI1 regulates differentiation, apoptosis and cell quiescence in leukemia and cell proliferation, migration, apoptosis and cell cycle in solid tumors.

Figure 4.
Downstream signaling pathways of EVI1. Known downstream signaling pathways of EVI1 include the PTEN/PI3K/AKT, TGF-β signaling, JNK and NF-kB pathways. The green bar represents DNA and the red arrow indicates the transcription start site.
Figure 4.
Downstream signaling pathways of EVI1. Known downstream signaling pathways of EVI1 include the PTEN/PI3K/AKT, TGF-β signaling, JNK and NF-kB pathways. The green bar represents DNA and the red arrow indicates the transcription start site.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liang, B.; Wang, J. EVI1 in Leukemia and Solid Tumors. Cancers 2020, 12, 2667. https://doi.org/10.3390/cancers12092667
AMA Style
Liang B, Wang J. EVI1 in Leukemia and Solid Tumors. Cancers. 2020; 12(9):2667. https://doi.org/10.3390/cancers12092667
Chicago/Turabian StyleLiang, Beiyuan, and Jing Wang. 2020. "EVI1 in Leukemia and Solid Tumors" Cancers 12, no. 9: 2667. https://doi.org/10.3390/cancers12092667
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.