RASSF1A Suppresses Estrogen-Dependent Breast Cancer Cell Growth through Inhibition of the Yes-Associated Protein 1 (YAP1), Inhibition of the Forkhead Box Protein M1 (FOXM1), and Activation of Forkhead Box Transcription Factor 3A (FOXO3A)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
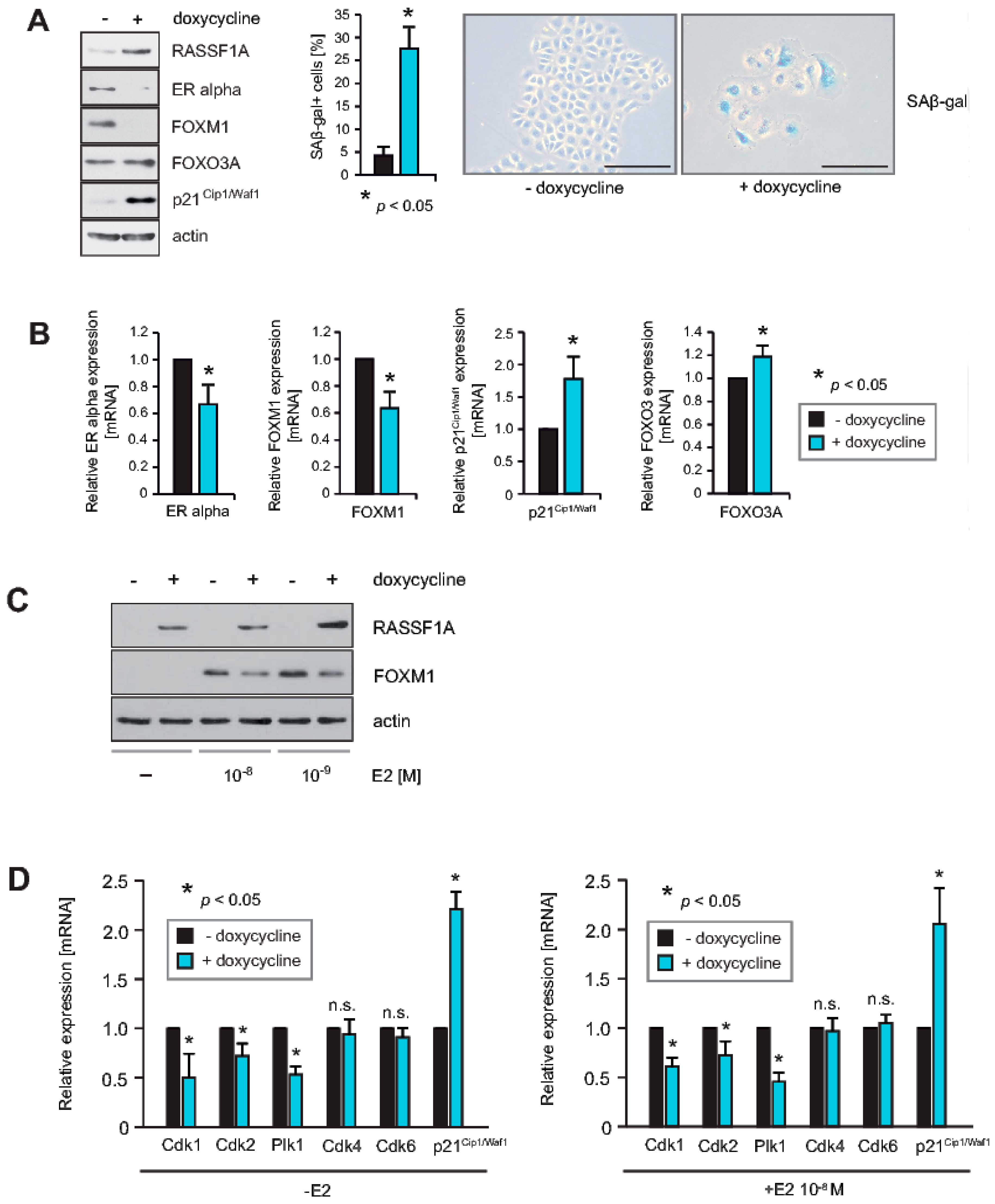
2.1. RASSF1A Causes Decreased Expression of FOXM1 and ERα, and Induces Senescence
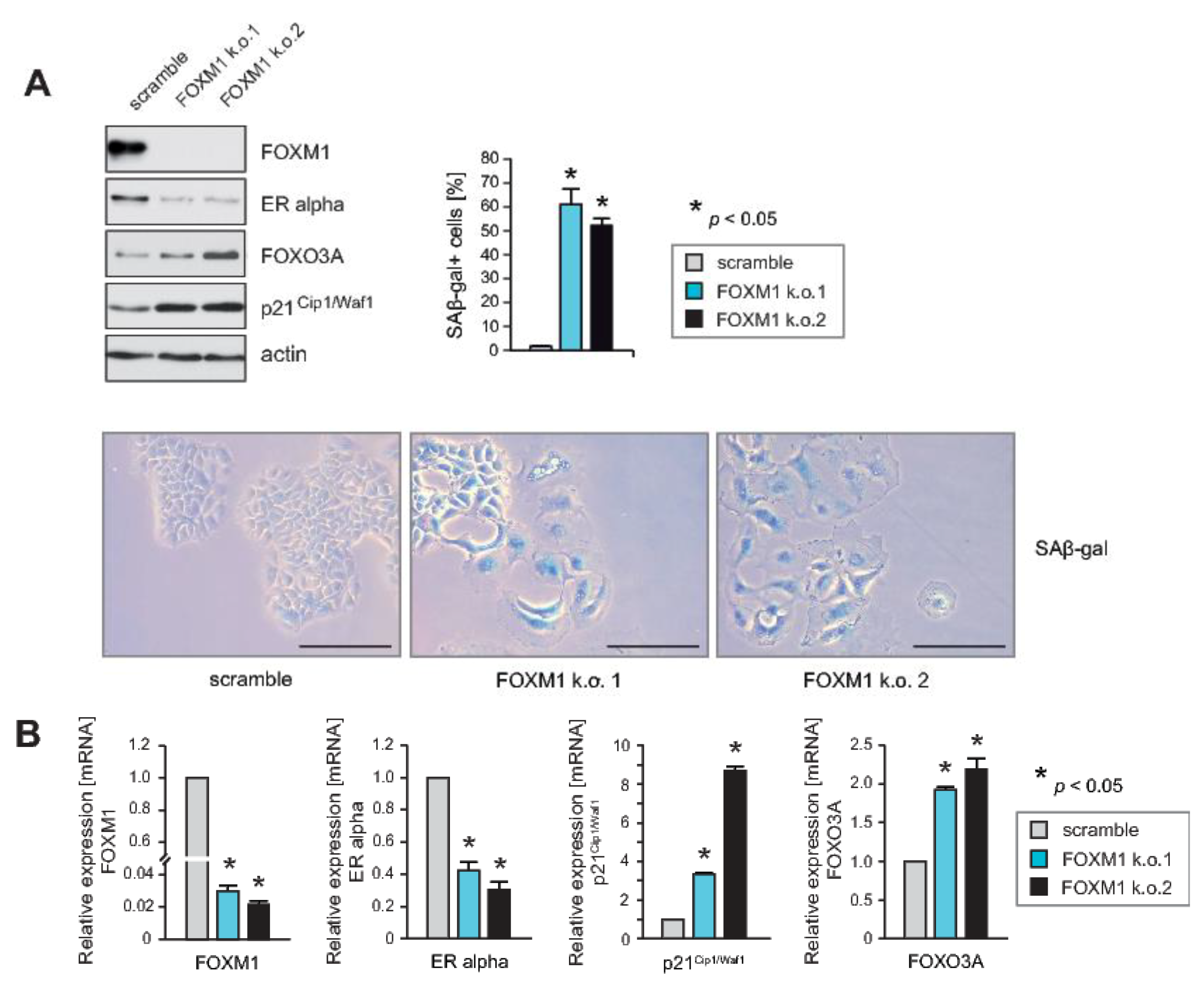
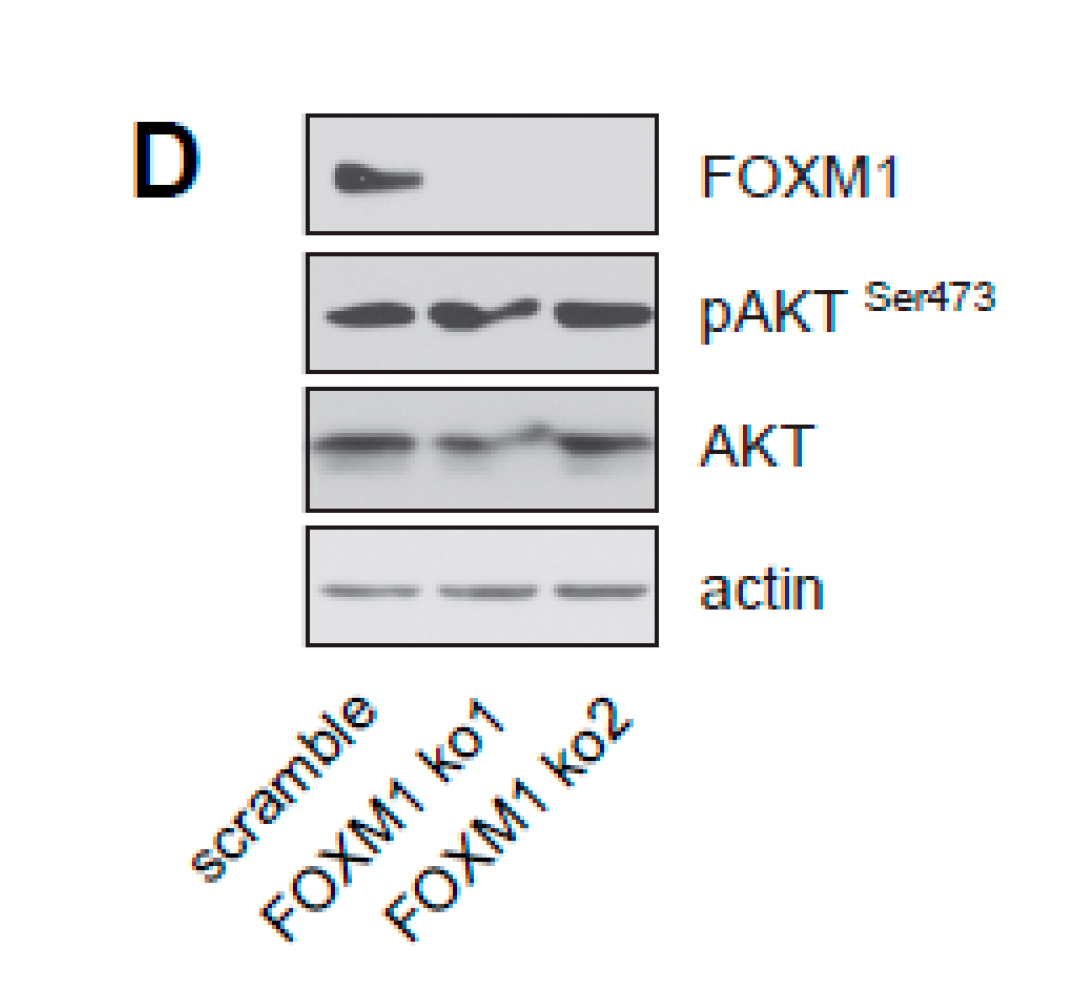
2.2. Knockdown of FOXM1 Inhibits Expression of ERα and Induces Cell Cycle Arrest and Senescence, Phenocopying the Effects of RASSF1A
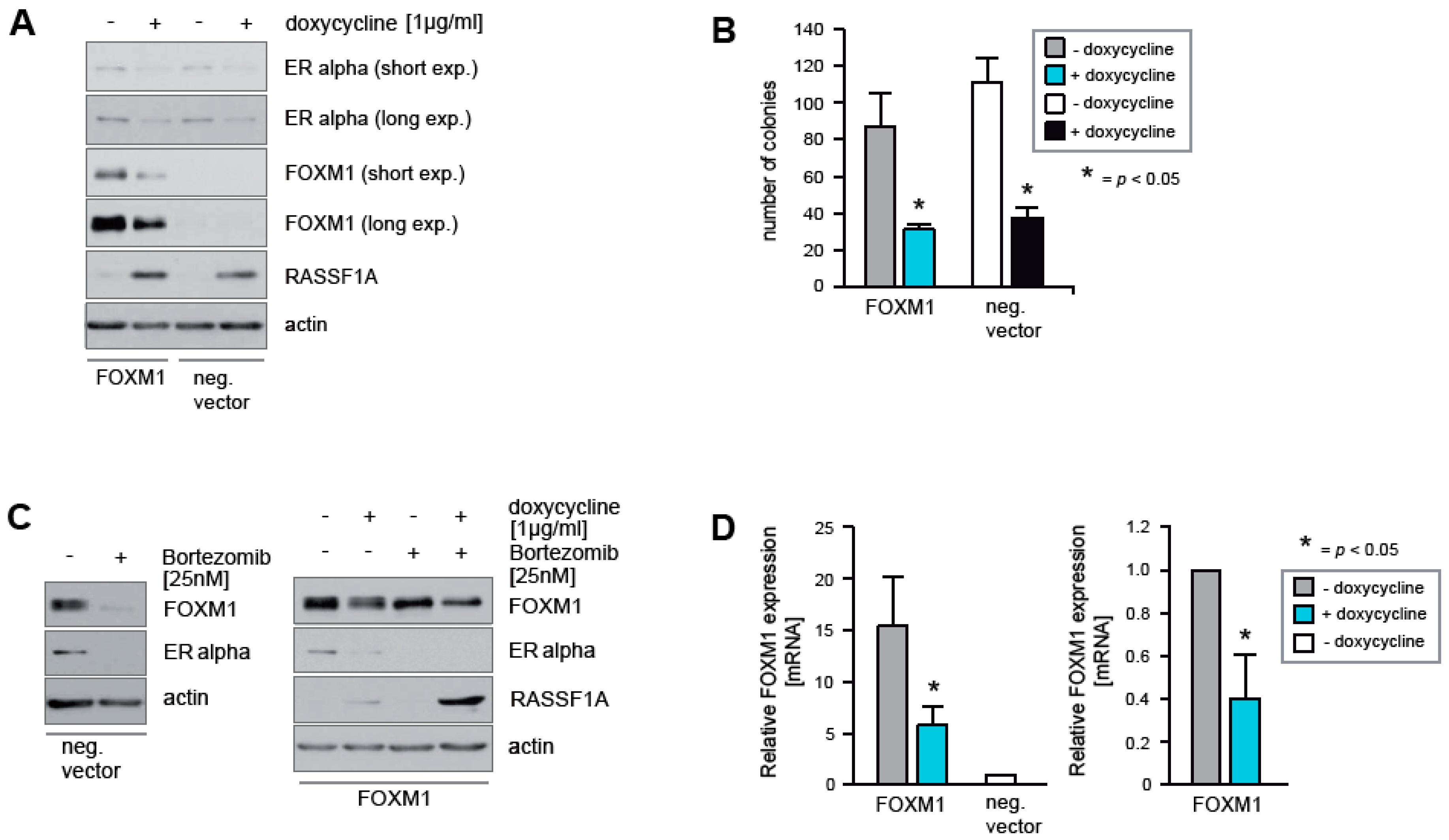
2.3. Ectopic Expression of FOXM1 Does Not Rescue Cells from RASSF1A-Mediated ERα Suppression and Cell Cycle Arrest
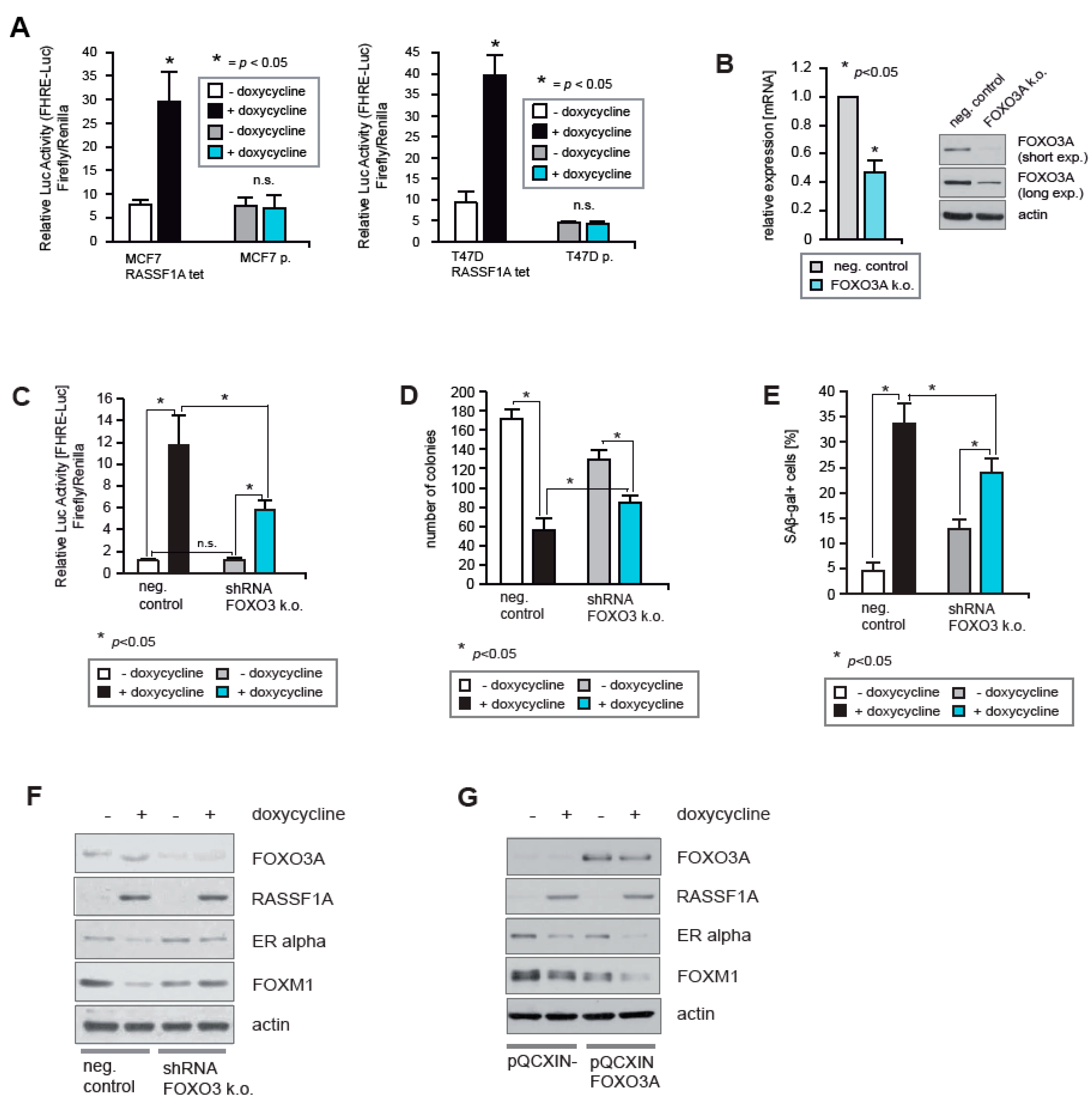
2.4. FOXO3A Is Required for RASSF1A-Mediated Growth Arrest
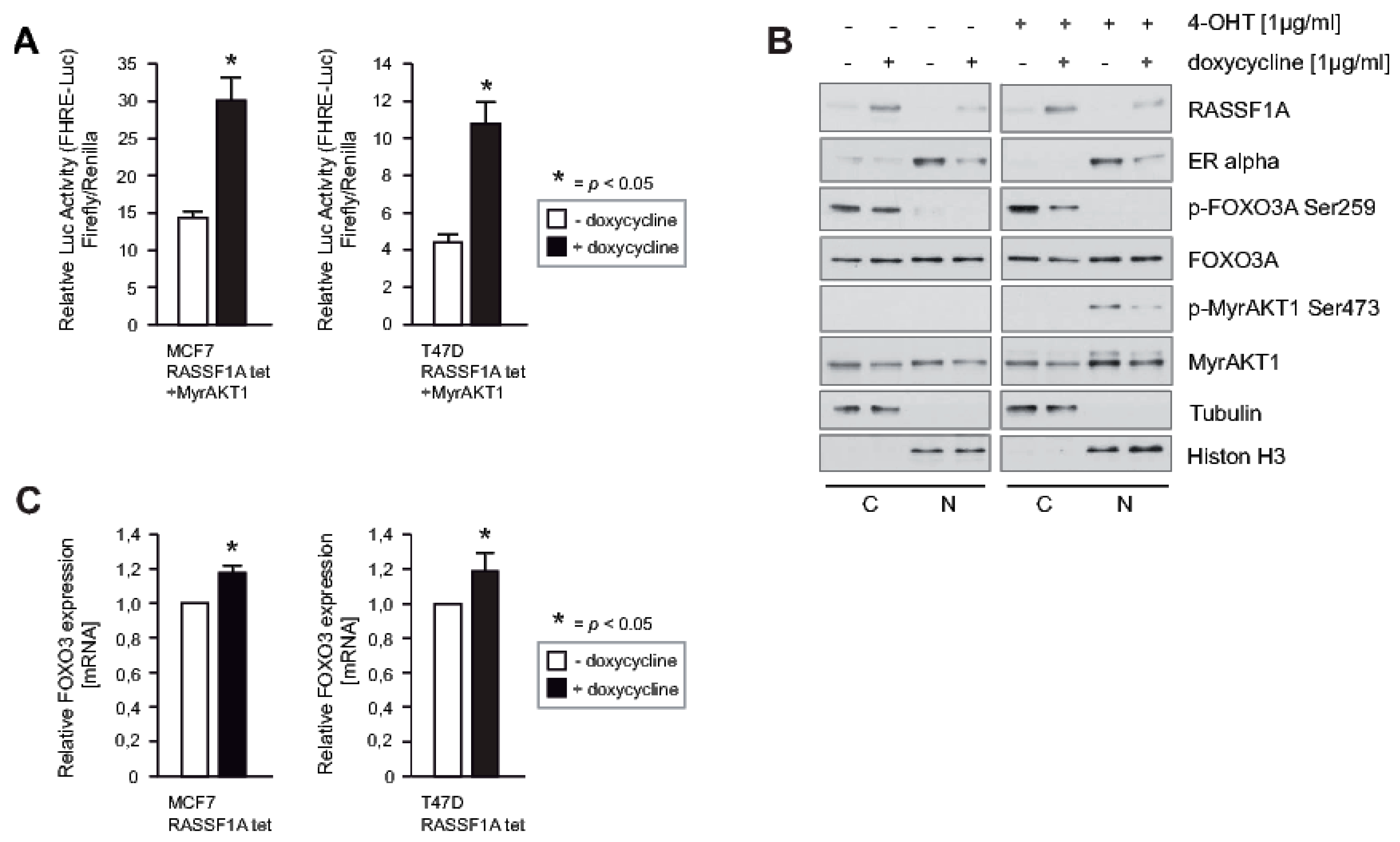
2.5. RASSF1A Increases FOXO3A Transcriptional Activity through Suppression of AKT-Mediated Inhibitory Phosphorylation and Increased Expression of FOXO3A
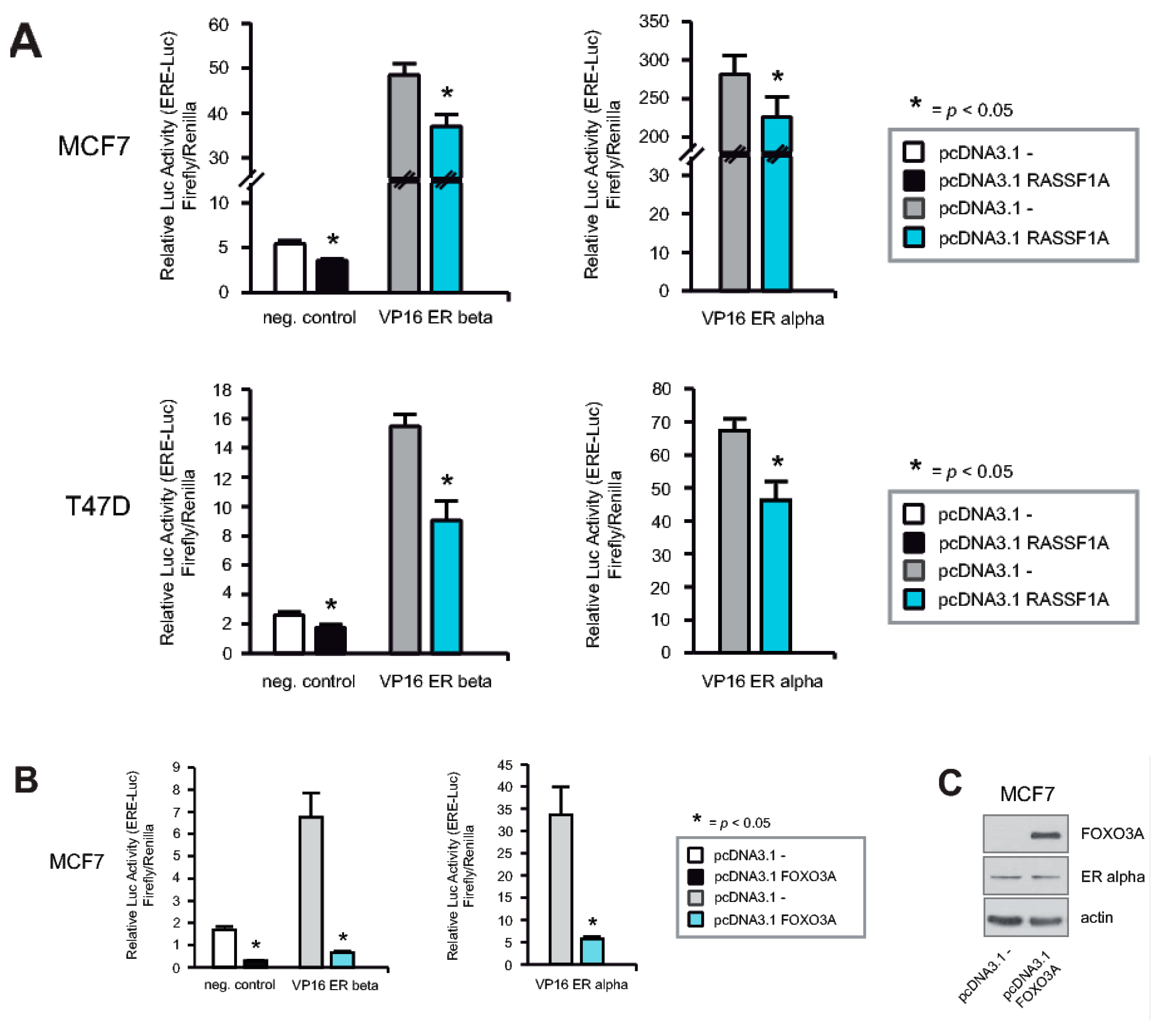
2.6. RASSF1A and FOXO3A Cause Decreased ERα and ERβ Activity
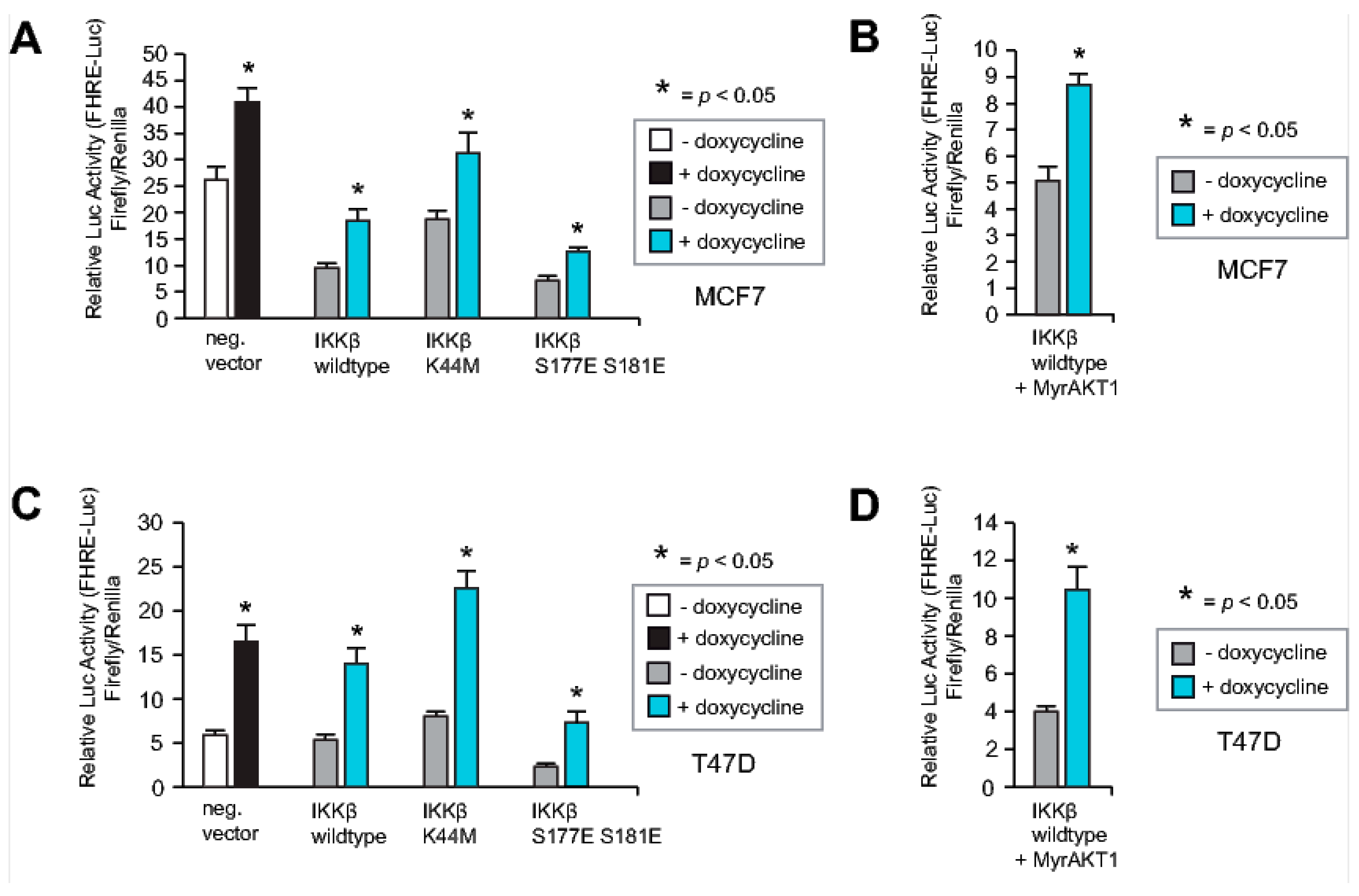
2.7. Neither IKKβ nor AKT1 Can Override the RASSF1A-Mediated Increase in FOXO3A Activity
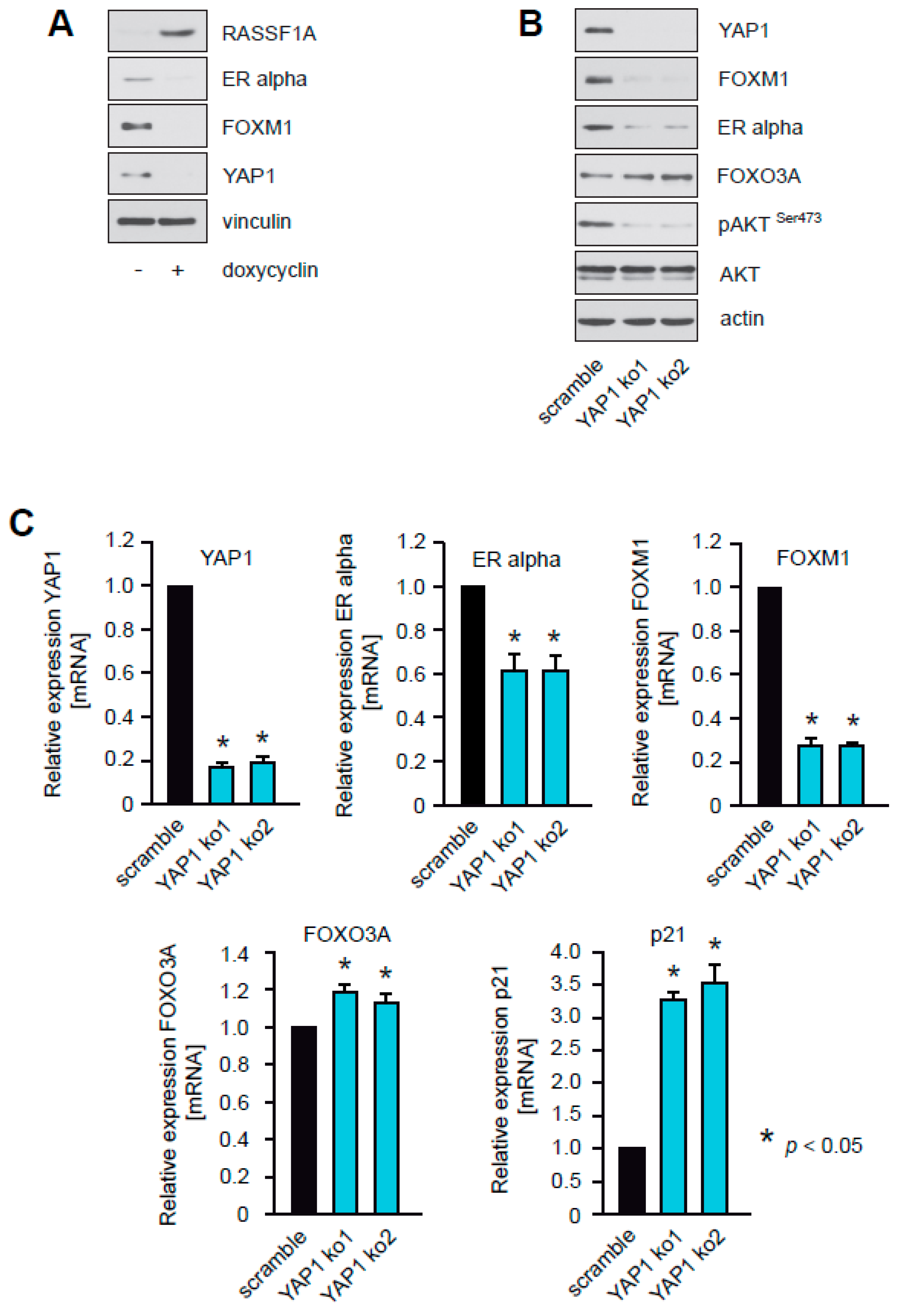
2.8. RASSF1A Inhibits YAP1, and Knockdown of YAP1 Suppresses Akt1 Activity, Inhibits Expression of ERα and FOXM1, and Increases Levels of FOXO3A
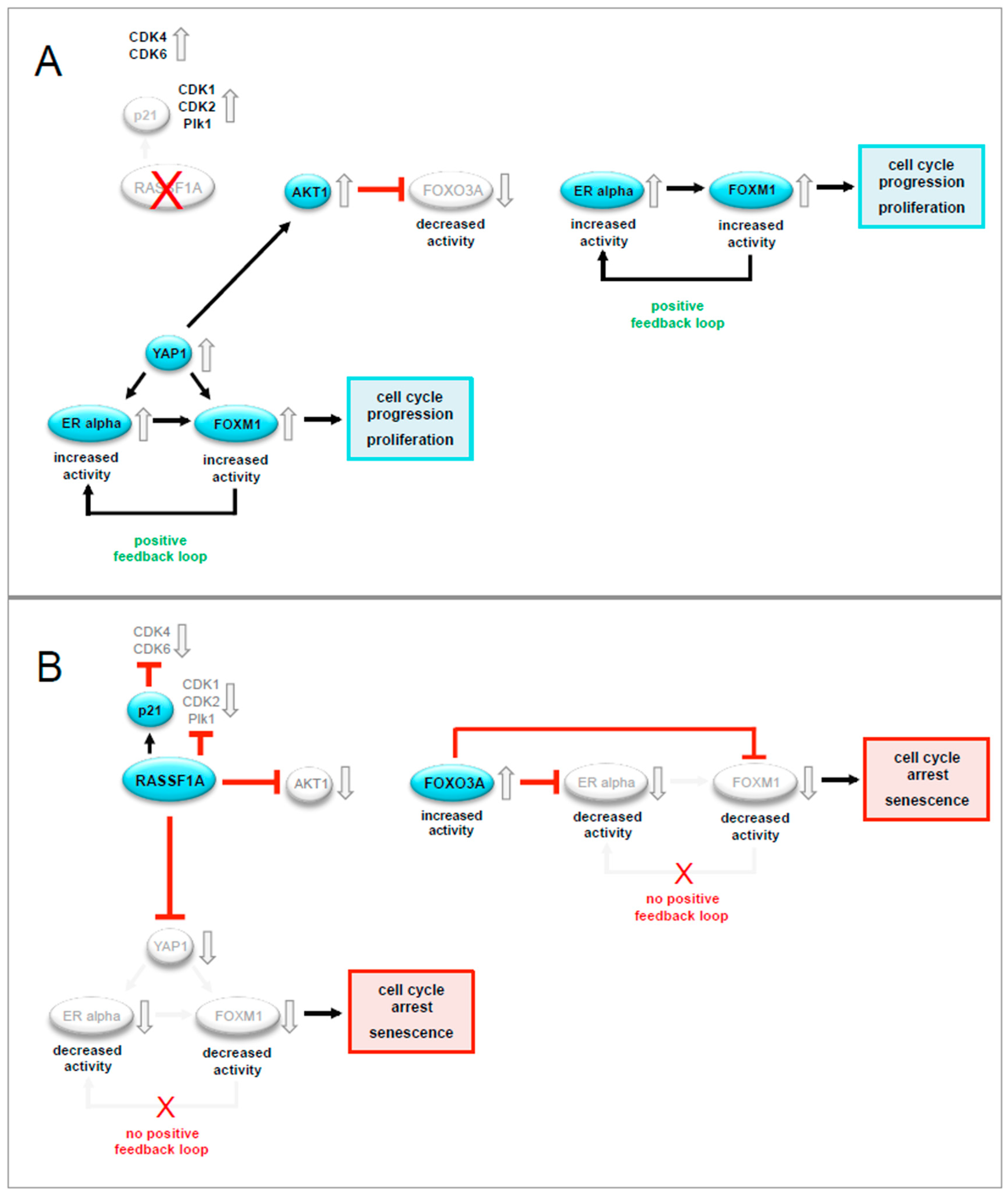
3. Discussion
4. Materials and Methods
4.1. Plasmids and Reagents
4.2. Cell Culture and Viral Transduction
4.3. Other Methods
4.4. Luciferase Assay Using the Dual Glow System (Promega)
4.5. Cell Cycle Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Keen, J.C.; Davidson, N.E. The biology of breast cancer. Cancer 2003, 97 (Suppl. 3), 825–833. [Google Scholar] [CrossRef]
- Thaler, S.; Schmidt, M.; Schad, A.; Sleeman, J.P. RASSF1A inhibits estrogen receptor alpha expression and estrogen-independent signalling: Implications for breast cancer development. Oncogene 2012, 31, 4912–4922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajabova, V.; Smolkova, B.; Zmetakova, I.; Sebova, K.; Krivulcik, T.; Bella, V.; Kajo, K.; Machalekova, K.; Fridrichova, I. RASSF1A promoter methylation levels positively correlate with estrogen receptor expression in breast cancer patients. Transl. Oncol. 2013, 6, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Plas, D.R.; Thompson, C.B. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J. Biol. Chem. 2003, 278, 12361–12366. [Google Scholar] [CrossRef] [Green Version]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell. Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.C.-T.; Lee, D.-F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IkB kinase promotes tumorigenesis through inhibition of Forkhead FOXO3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Regan, K.M.; Wang, F.; Wang, D.; Smith, D.I.; van Deursen, J.M.; Tindall, D.J. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lethinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villen, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.K.; McGuire, V.A.; Koo, C.Y.; Muir, K.W.; de Olano, N.; Maifoshie, E.; Kelly, D.J.; McGovern, U.B.; Monteiro, L.J.; Gomes, A.R.; et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J. Biol. Chem. 2012, 287, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer. 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Khongkow, P.; Karunarathna, U.; Khongkow, M.; Gong, C.; Gomes, A.R.; Yagüe, E.; Monteiro, L.J.; Kongsema, M.; Zone, S.; Man, E.P.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2014, 33, 4144–4155. [Google Scholar] [CrossRef] [Green Version]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Millour, J.; Constantinidou, D.; Stavropoulou, A.V.; Wilson, M.S.C.; Myatt, S.S.; Kwok, J.M.; Sivanandan, K.; Coombes, R.C.; Medema, R.H.; Hartman, J.; et al. FOXM1 is a transcriptional target of the ERα and has a critical role in breast cancer endocrine sensitivity and resistance. Oncogene 2010, 29, 2983–2995. [Google Scholar] [CrossRef] [Green Version]
- Madureira, P.A.; Varshochi, R.; Constantinidou, D.; Francis, R.E.; Coombes, R.C.; Yao, K.M.; Lam, E.W. The Forkhead box M1 protein regulates the transcription of the estrogen receptor alpha in breast cancer cells. J. Biol. Chem. 2006, 281, 25167–25176. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Tsai, W.B.; Cheng, C.J.; Hsu, C.; Min, C.Y.; Li, P.C.; Lin, S.H.; Hu, M.C.T. Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res. 2007, 10, R21–R34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, C.Y.; Muir, K.W.; Lam, E.W. FOXM1: From cancer initiation to progression and treatment. Biochim. Biophys. Acta 2012, 1819, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Thaler, S.; Hähnel, P.S.; Schad, A.; Dammann, R.; Schuler, M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009, 69, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittfeld, C.; Richter, A.M.; Steinmann, K.; Klagge-Ulonska, A.; Dammann, R.H. The SARAH domain of RASSF1A and ist tumor suppressor function. Mol. Biol. Int. 2012, 2012, 196715. [Google Scholar] [CrossRef] [PubMed]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef]
- Jimenez, A.P.; Traum, A.; Boettger, T.; Hackstein, H.; Richter, A.M.; Dammann, R.H. The tumor suppressor RASSF1A induces the YAP1 target gene ANKRD1 that is epigenetically inactivated in human cancers and inhibits tumor growth. Oncotarget 2017, 8, 88437–88452. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, Q.; Liu, Q.; Li, Y.; Sun, X.; Hong, L.; Ji, S.; Liu, C.; Geng, J.; Zhang, W.; et al. Hippo signaling suppresses cell ploidy and tumorigenesis through Skp2. Cancer Cell 2017, 31, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Dominguez-Brauer, C.; Wang, Z.; Asara, J.M.; Costa, R.H.; Tyner, A.L.; Lau, L.F.; Raychaudhuri, P. A conserved phosphorylation site within the forkhead domain of FoxM1B is required for its activation by cyclin-CDK1. J. Biol. Chem. 2009, 284, 30695–30707. [Google Scholar] [CrossRef] [Green Version]
- Wierstra, I.; Alves, J. Transcription factor FOXM1c is repressed by RB and activated by cyclin D1/Cdk4. Biol. Chem. 2006, 387, 949–962. [Google Scholar] [CrossRef]
- Laoukili, J.; Alvarez, M.; Meijer, L.A.; Stahl, M.; Mohammed, S.; Kleij, L.; Heck, A.J.; Medema, R.H. Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autosuppression by the FoxM1 N-terminal domain. Mol. Cell Biol. 2008, 28, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Malureanu, L.; Huang, J.; Wang, W.; Li, H.; van Deursen, J.M.; Tindall, D.J.; Chen, J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol. 2008, 10, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cycline-dependent kinases by p21. Mol. Biol. Cell. 1995, 4, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, S.; Thiede, G.; Hengstler, J.G.; Schad, A.; Schmidt, M.; Sleeman, J.P. The proteasome inhibitor Bortezomib (Velcade) as potential inhibitor of estrogen receptor-positive breast cancer. Int. J. Cancer 2015, 137, 686–687. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Nakamura, N.; Vazquez, F.; Batt, D.B.; Perera, S.; Roberts, T.M.; Sellers, W.R. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 2110–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, A.D.; Barthel, A.; Kovacina, K.S.; Boge, A.; Wallach, B.; Summers, S.A.; Birnbaum, M.J.; Scott, P.H.; Lawrence, J.C., Jr.; Roth, R.A. Construction and characterization of a conditionally active version of the serine/threonine kinase Akt. J. Biol. Chem. 1998, 273, 11937–11943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.; Norris, J.D.; Gron, H.; Paige, L.A.; Hamilton, P.T.; Kenan, D.J.; Fowlkes, D.; McDonnell, D.P. Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libaries: Discovery of peptide antagonists of estrogen receptors alpha and beta. Mol. Cell. Biol. 1999, 19, 8226–8239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.M.; McDonnell, D.P. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 1999, 140, 5566–5578. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, F.; Zhu, H.; Murray, B.W.; Shevchenko, A.; Bennett, B.L.; Li, J.; Young, D.B.; Barbosa, M.; Mann, M.; Manning, A.; et al. IKK-1 and IKK-2: Cytokine-activated IkappaB kinase essential for NF-kappaB activation. Science 1997, 278, 860–866. [Google Scholar] [CrossRef]
- Dammann, R.; Schadarsurengin, U.; Seidel, C.; Strunnikova, M.; Rastetter, M.; Baier, K.; Pfeifer, G.P. The tumor suppressor RASSF1A in human carcinogenesis: An update. Histol. Histopathol. 2005, 20, 645–663. [Google Scholar]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amanti, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zeng, J.; Zhou, M.; Li, B.; Zhang, Y.; Huang, T.; Wang, L.; Jia, J.; Chen, C. The tumor suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid leukemia. Mol. Cancer 2012, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Chen, W.; Miao, R.; Zhou, Y.; Wang, Z.; Zhang, L.; Wan, Y.; Dong, Y.; Qu, K.; Liu, C. miR-34a induces cellular senescence via modulation of telomerase activity in human hepatocellular carcinoma by targeting FoxM1/c-Myc pathway. Oncotarget 2015, 6, 3988–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, J.R.; Kiefer, M.M.; Park, H.J.; Li, J.; Wang, Z.; Fontanarosa, J.; DeWaal, D.; Kopanja, D.; Benevolenskaya, E.V.; Guzman, G.; et al. FoxM1 regulates mammary luminal cell fate. Cell Rep. 2012, 1, 715–729. [Google Scholar] [CrossRef] [Green Version]
- Teh, M.T.; Gemenetzides, E.; Patel, D.; Tariq, R.; Nadir, A.; Batha, A.W.; Waseem, A.; Hutchison, I.L. FOXM1 induces a global methylation signature that mimics the cancer epigenome in head and neck squamous cell carcinoma. PLoS ONE 2012, 7, e34329. [Google Scholar] [CrossRef] [PubMed]
- Palakurthy, R.K.; Wajapeyee, N.; Santra, M.K.; Gazin, C.; Lin, L.; Gobeil, S.; Green, M.R. Epigenetic silencing of the RASSF1A tumor suppressor gene through HOXB3-mediated induction of DNMT3B expression. Mol. Cell. 2009, 36, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Zhang, X.; Pfeifer, G.P. The tumor suppressor rassf1a prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J. Biol. Chem. 2011, 268, 6253–6261. [Google Scholar] [CrossRef] [Green Version]
- Pefani, D.E.; Latusek, R.; Pires, I.; Grawenda, A.M.; Yee, K.S.; Hamilton, G.; van der Weyden, L.; Esashi, F.; Hammond, E.M.; O’Neill, E. RASSF1A-LATS1 signalling stabilizes replication forks by restricting CDK2-mediated phosphorylation of BRCA2. Nat. Cell. Biol. 2014, 16, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Yang, J.; Chen, X.; Li, J.; Li, X.; Wang, L.; Tan, Y.; Xiong, W.; Zhou, M.; McCarthy, J.B.; et al. RASSF1A suppresses melanoma development by modulating apoptosis and cell-cycle progression. J. Cell Physiol. 2011, 226, 2360–2369. [Google Scholar] [CrossRef] [Green Version]
- Bucur, O.; Stancu, A.L.; Muraru, M.S.; Melet, A.; Petrescu, S.M.; Khosravi-Far, R. PLK1 is a binding partner and a negative regulator of FOXO3 tumor suppressor. Discoveries 2014, 2, e16. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roßwag, S.; Thiede, G.; Sleeman, J.P.; Thaler, S. RASSF1A Suppresses Estrogen-Dependent Breast Cancer Cell Growth through Inhibition of the Yes-Associated Protein 1 (YAP1), Inhibition of the Forkhead Box Protein M1 (FOXM1), and Activation of Forkhead Box Transcription Factor 3A (FOXO3A). Cancers 2020, 12, 2689. https://doi.org/10.3390/cancers12092689
Roßwag S, Thiede G, Sleeman JP, Thaler S. RASSF1A Suppresses Estrogen-Dependent Breast Cancer Cell Growth through Inhibition of the Yes-Associated Protein 1 (YAP1), Inhibition of the Forkhead Box Protein M1 (FOXM1), and Activation of Forkhead Box Transcription Factor 3A (FOXO3A). Cancers. 2020; 12(9):2689. https://doi.org/10.3390/cancers12092689
Chicago/Turabian StyleRoßwag, Sven, Gitta Thiede, Jonathan P. Sleeman, and Sonja Thaler. 2020. "RASSF1A Suppresses Estrogen-Dependent Breast Cancer Cell Growth through Inhibition of the Yes-Associated Protein 1 (YAP1), Inhibition of the Forkhead Box Protein M1 (FOXM1), and Activation of Forkhead Box Transcription Factor 3A (FOXO3A)" Cancers 12, no. 9: 2689. https://doi.org/10.3390/cancers12092689