High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
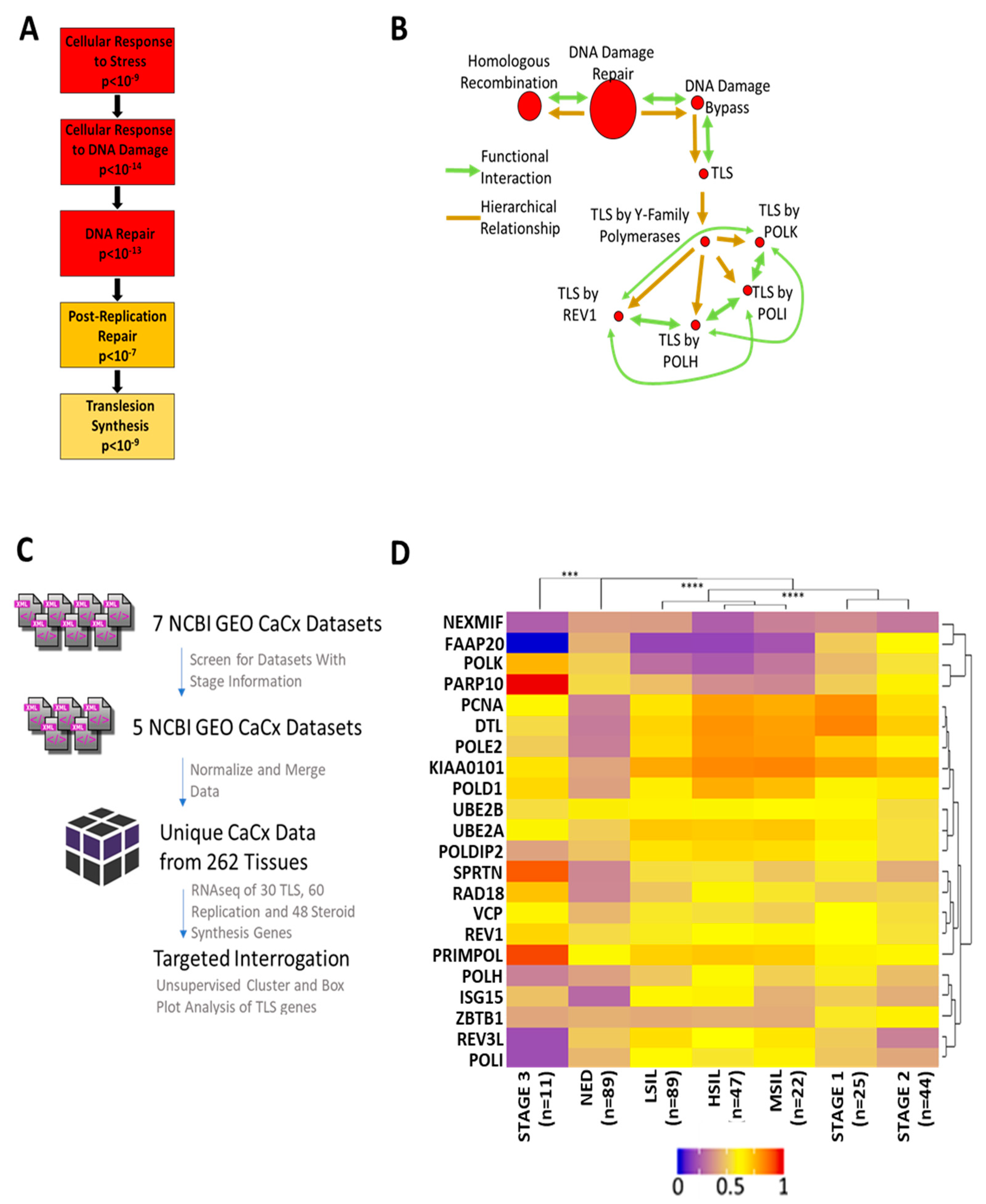
2.1. Transcriptomic Analysis Reveals Altered Translesion Synthesis Gene Expression in CaCx
2.2. In Vitro and Ex Vivo Systems Confirm Dysregulated TLS Gene Expression in CaCx
2.3. HPV Oncogenes Disrupt TLS Polymerase Eta Induction
2.4. HPV E6 Blocks POLη Induction in a p53-Dependent Manner
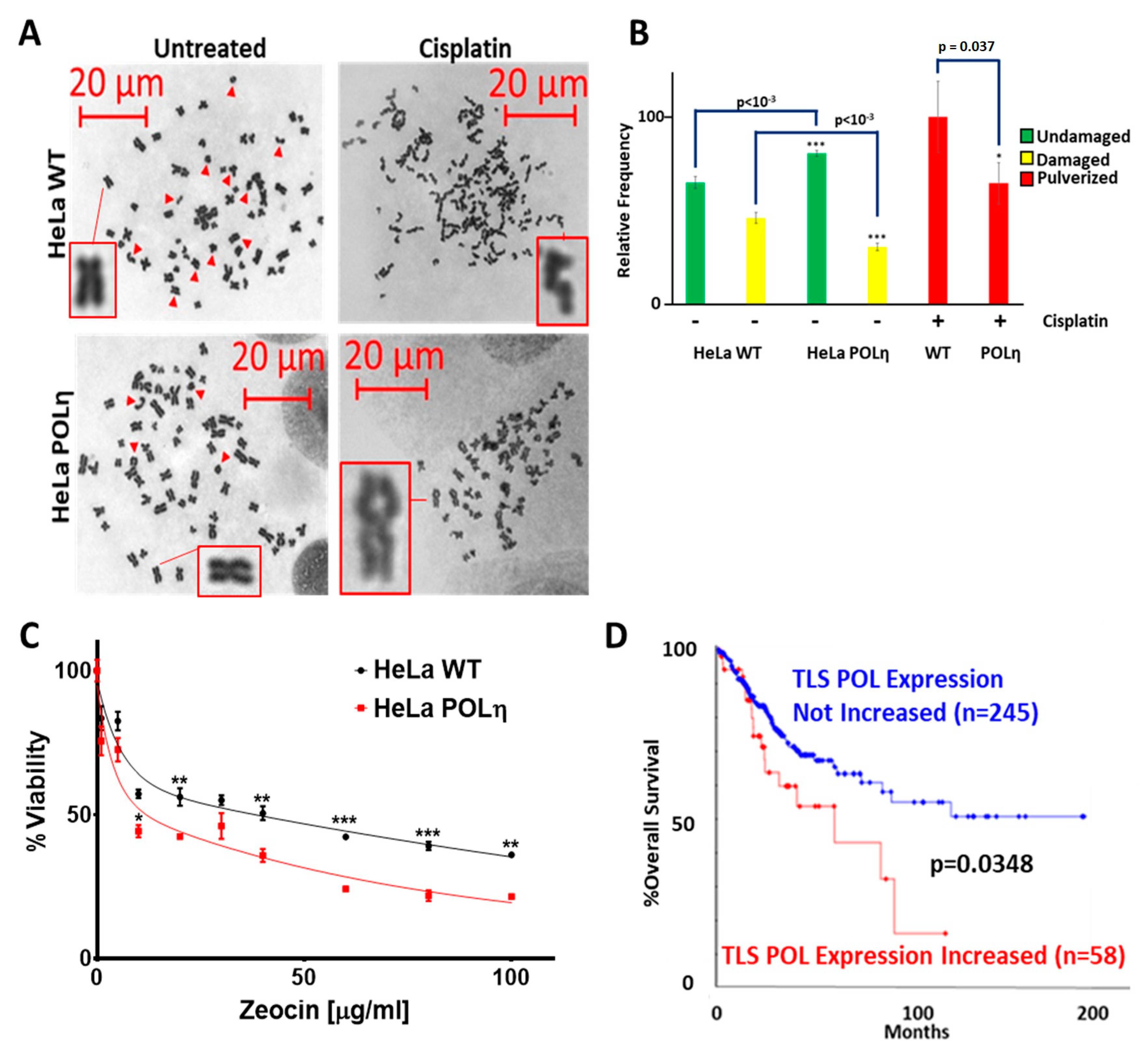
2.5. The Inability to Induce POLη Contributes to Cisplatin Sensitivity in Cervical Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Antibodies and Chemicals
4.2. Pathway-by-Stage and Gene Ontology
4.3. Cbioportal Analysis of TCGA Data
4.4. Quantitative RT-PCR
4.5. UV-Timeseries and POLη Induction
4.6. HeLa/SiHa Nucleoside Addition Experiment
4.7. TLS Protein Stability
4.8. Cell Culture
- HeLa (ATCC® CCL-2™)
- SiHa (ATCC® HTB-35™)
- C-33 A [c-33a] (ATCC® HTB-31™)
- HFK
4.9. UV and DNA-Damaging Agent Sensitivity
4.10. Damaged Chromosomes Detection by Metaphase Spread
4.11. Immunofluorescence Microscopy
4.12. Pathology Analysis
4.13. Statistical Analysis
4.14. E6 F47R Transient Transfections
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.-M. Classification of Papillomaviruses (PVs) Based on 189 PV Types and Proposal of Taxonomic Amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorslaer, K.; Burk, R.D. Evolution of Human Papillomavirus Carcinogenicity. Adv. Virus Res. 2010, 77, 41–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, E.M.; Talpe-Nunes, V.; Sichero, L. Epidemiology and biology of cutaneous human papillomavirus. Clinics 2018, 73, e489s. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Doorbar, J. The low-risk papillomaviruses. Virus Res. 2017, 231, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, M.; De Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global burden of cancers attributable to infections in 2012: A synthetic analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef] [Green Version]
- Moscicki, A.-B.; Palefsky, J.M. HPV in men: An update. J. Low. Genit. Tract. Dis. 2011, 15, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Vanthoor, J.; Vos, G.; Albersen, M. Penile cancer: Potential target for immunotherapy? World J. Urol. 2020. [Google Scholar] [CrossRef]
- Preti, M.; Rotondo, J.C.; Holzinger, D.; Micheletti, L.; Gallio, N.; McKay-Chopin, S.; Carreira, C.; Privitera, S.S.; Watanabe, R.; Ridder, R.; et al. Role of human papillomavirus infection in the etiology of vulvar cancer in Italian women. Infect Agent Cancer 2020, 15, 20. [Google Scholar] [CrossRef] [Green Version]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, N.; Guida, P.; Münger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Khanal, S.; Galloway, D.A. High-risk human papillomavirus oncogenes disrupt the Fanconi anemia DNA repair pathway by impairing localization and de-ubiquitination of FancD2. PLoS Pathog. 2019, 15, e1007442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, N.A. Catching HPV in the Homologous Recombination Cookie Jar. Trends Microbiol. 2020, 28, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Soto, D.R.; Barton, C.; Munger, K.; McLaughlin-Drubin, M.E. KDM6A addiction of cervical carcinoma cell lines is triggered by E7 and mediated by p21CIP1 suppression of replication stress. PLOS Pathog. 2017, 13, e1006661. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Moody, C.A. Impact of Replication Stress in Human Papillomavirus Pathogenesis. J. Virol. 2019, 93, e01012-17. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Acharya, N.; Yoon, J.-H.; Gali, H.; Unk, I.; Haracska, L.; Johnson, R.E.; Hurwitz, J.; Prakash, L.; Prakash, S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase η in translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 17724–17729. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Biertümpfel, C.; Gregory, M.T.; Hua, Y.-J.; Hanaoka, F.; Yang, W. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent Cisplatin-Based Radiotherapy and Chemotherapy for Locally Advanced Cervical Cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Alberts, D.S.; Garcia, D.; Mason-Liddil, N. Cisplatin in advanced cancer of the cervix: An update. Semin. Oncol. 1991, 18, 11–24. [Google Scholar]
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [Green Version]
- Guardado-Estrada, M.; Medina-Martínez, I.; Juárez-Torres, E.; Roman-Bassaure, E.; Macías, L.; Alfaro, A.; Alcántara-Vázquez, A.; Alonso, P.; Gomez, G.; Cruz-Talonia, F.; et al. The Amerindian mtDNA haplogroup B2 enhances the risk of HPV for cervical cancer: De-regulation of mitochondrial genes may be involved. J. Hum. Genet. 2012, 57, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Pyeon, D.; Newton, M.A.; Lambert, P.F.; den Boon, J.A.; Sengupta, S.; Marsit, C.J.; Woodworth, C.D.; Connor, J.P.; Haugen, T.H.; Smith, E.M.; et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007, 67, 4605–4619. [Google Scholar] [CrossRef] [Green Version]
- Scotto, L.; Narayan, G.; Nandula, S.V.; Arias-Pulido, H.; Subramaniyam, S.; Schneider, A.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes Chromosomes Cancer 2008, 47, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Kuick, R.; Nan, B.; Ota, I.; Weiss, S.J.; Trimble, C.L.; Fearon, E.R.; Cho, K.R. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007, 67, 10163–10172. [Google Scholar] [CrossRef] [Green Version]
- Cruet-Hennequart, S.; Coyne, S.; Glynn, M.T.; Oakley, G.G.; Carty, M.P. UV-induced RPA phosphorylation is increased in the absence of DNA polymerase eta and requires DNA-PK. DNA Repair 2006, 5, 491–504. [Google Scholar] [CrossRef]
- Sokka, M.; Parkkinen, S.; Pospiech, H.; Syväoja, J.E. Function of TopBP1 in genome stability. Subcell. Biochem. 2010, 50, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The Rb binding domain of HPV31 E7 is required to maintain high levels of DNA repair factors in infected cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.-E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [Green Version]
- Essers, J.; Theil, A.F.; Baldeyron, C.; van Cappellen, W.A.; Houtsmuller, A.B.; Kanaar, R.; Vermeulen, W. Nuclear Dynamics of PCNA in DNA Replication and Repair. Mol. Cell Biol. 2005, 25, 9350–9359. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X. DNA polymerase eta, the product of the xeroderma pigmentosum variant gene and a target of p53, modulates the DNA damage checkpoint and p53 activation. Mol. Cell. Biol. 2006, 26, 1398–1413. [Google Scholar] [CrossRef] [Green Version]
- Lerner, L.K.; Francisco, G.; Soltys, D.T.; Rocha, C.R.R.; Quinet, A.; Vessoni, A.T.; Castro, L.P.; David, T.I.P.; Bustos, S.O.; Strauss, B.E.; et al. Predominant role of DNA polymerase eta and p53-dependent translesion synthesis in the survival of ultraviolet-irradiated human cells. Nucleic Acids Res. 2017, 45, 1270–1280. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Pebernard, S.; Iggo, R.D. Regulation of p53 Stability and Function in HCT116 Colon Cancer Cells. J. Biol. Chem. 2004, 279, 7598–7605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristriani, T.; Fournane, S.; Orfanoudakis, G.; Travé, G.; Masson, M. A single-codon mutation converts HPV16 E6 oncoprotein into a potential tumor suppressor, which induces p53-dependent senescence of HPV-positive HeLa cervical cancer cells. Oncogene 2009, 28, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertella, M.R.; Green, C.M.; Lehmann, A.R.; O’Connor, M.J. A role for polymerase eta in the cellular tolerance to cisplatin-induced damage. Cancer Res. 2005, 65, 9799–9806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatraman, M.; Anto, R.J.; Nair, A.; Varghese, M.; Karunagaran, D. Biological and chemical inhibitors of NF-kappaB sensitize SiHa cells to cisplatin-induced apoptosis. Mol. Carcinog. 2005, 44, 51–59. [Google Scholar] [CrossRef]
- Bertoletti, F.; Cea, V.; Liang, C.-C.; Lanati, T.; Maffia, A.; Avarello, M.D.M.; Cipolla, L.; Lehmann, A.R.; Cohn, M.A.; Sabbioneda, S. Phosphorylation regulates human polη stability and damage bypass throughout the cell cycle. Nucleic Acids Res. 2017, 45, 9441–9454. [Google Scholar] [CrossRef]
- Yang, L.; Shi, T.; Liu, F.; Ren, C.; Wang, Z.; Li, Y.; Tu, X.; Yang, G.; Cheng, X. REV3L, a Promising Target in Regulating the Chemosensitivity of Cervical Cancer Cells. PLoS ONE 2015, 10, e0120334. [Google Scholar] [CrossRef]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small Interfering RNA Screens Reveal Enhanced Cisplatin Cytotoxicity in Tumor Cells Having both BRCA Network and TP53 Disruptions. Mol. Cell. Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.-Y.; Yang, L.; Yang, G.; Tu, X.-Y.; Wu, X.; Cheng, X.; Wei, Q. DNA polymerase ζ as a potential biomarker of chemoradiation resistance and poor prognosis for cervical cancer. Med. Oncol. 2013, 30, 500. [Google Scholar] [CrossRef]
- Long, H.J. Management of Metastatic Cervical Cancer: Review of the Literature. JCO 2007, 25, 2966–2974. [Google Scholar] [CrossRef]
- Fischer, M.; Uxa, S.; Stanko, C.; Magin, T.M.; Engeland, K. Human papilloma virus E7 oncoprotein abrogates the p53-p21-DREAM pathway. Sci. Rep. 2017, 7, 2603. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Petrelli, F.; Coinu, A.; Raspagliesi, F.; Barni, S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol. Oncol. 2014, 133, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Sill, M.W.; McMeekin, D.S.; Cohn, D.E.; Ramondetta, L.M.; Boardman, C.H.; Benda, J.; Cella, D. Phase III trial of four cisplatin-containing doublet combinations in stage IVB, recurrent, or persistent cervical carcinoma: A Gynecologic Oncology Group study. J. Clin. Oncol. 2009, 27, 4649–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, K.S.; Sill, M.W.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; Michael, H.E.; et al. Bevacizumab for advanced cervical cancer: Final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240). Lancet 2017, 390, 1654–1663. [Google Scholar] [CrossRef] [Green Version]
- Vogl, S.E.; Seltzer, V.; Camacho, F.; Calanog, A. Dianhydrogalactitol and cisplatin in combination for advanced cancer of the uterine cervix. Cancer Treat. Rep. 1982, 66, 1809–1812. [Google Scholar]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Devel. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef] [Green Version]
- Stillman, B. Smart machines at the DNA replication fork. Cell 1994, 78, 725–728. [Google Scholar] [CrossRef]
- Johnson, R.E.; Washington, M.T.; Prakash, S.; Prakash, L. Bridging the gap: A family of novel DNA polymerases that replicate faulty DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 12224–12226. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.F.; Lin, H.K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar]
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank–rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [Green Version]
- Eden, E.; Lipson, D.; Yogev, S.; Yakhini, Z. Discovering Motifs in Ranked Lists of DNA Sequences. PLOS Comput. Biol. 2007, 3, e39. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Howe, B.; Umrigar, A.; Tsien, F. Chromosome Preparation from Cultured Cells. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [Green Version]
- Murthy, V.; Dacus, D.; Gamez, M.; Hu, C.; Wendel, S.O.; Snow, J.; Kahn, A.; Walterhouse, S.H.; Wallace, N.A. Characterizing DNA Repair Processes at Transient and Long-lasting Double-strand DNA Breaks by Immunofluorescence Microscopy. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wendel, S.O.; Snow, J.A.; Bastian, T.; Brown, L.; Hernandez, C.; Burghardt, E.; Kahn, A.; Murthy, V.; Neill, D.; Smith, Z.C.; et al. High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction. Cancers 2021, 13, 28. https://doi.org/10.3390/cancers13010028
Wendel SO, Snow JA, Bastian T, Brown L, Hernandez C, Burghardt E, Kahn A, Murthy V, Neill D, Smith ZC, et al. High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction. Cancers. 2021; 13(1):28. https://doi.org/10.3390/cancers13010028
Chicago/Turabian StyleWendel, Sebastian O., Jazmine A. Snow, Tyler Bastian, Laura Brown, Candy Hernandez, Emily Burghardt, Andrew Kahn, Vaibhav Murthy, Daniel Neill, Zachary C. Smith, and et al. 2021. "High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction" Cancers 13, no. 1: 28. https://doi.org/10.3390/cancers13010028