
KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas


Abstract
:Simple Summary
Abstract
1. Introduction
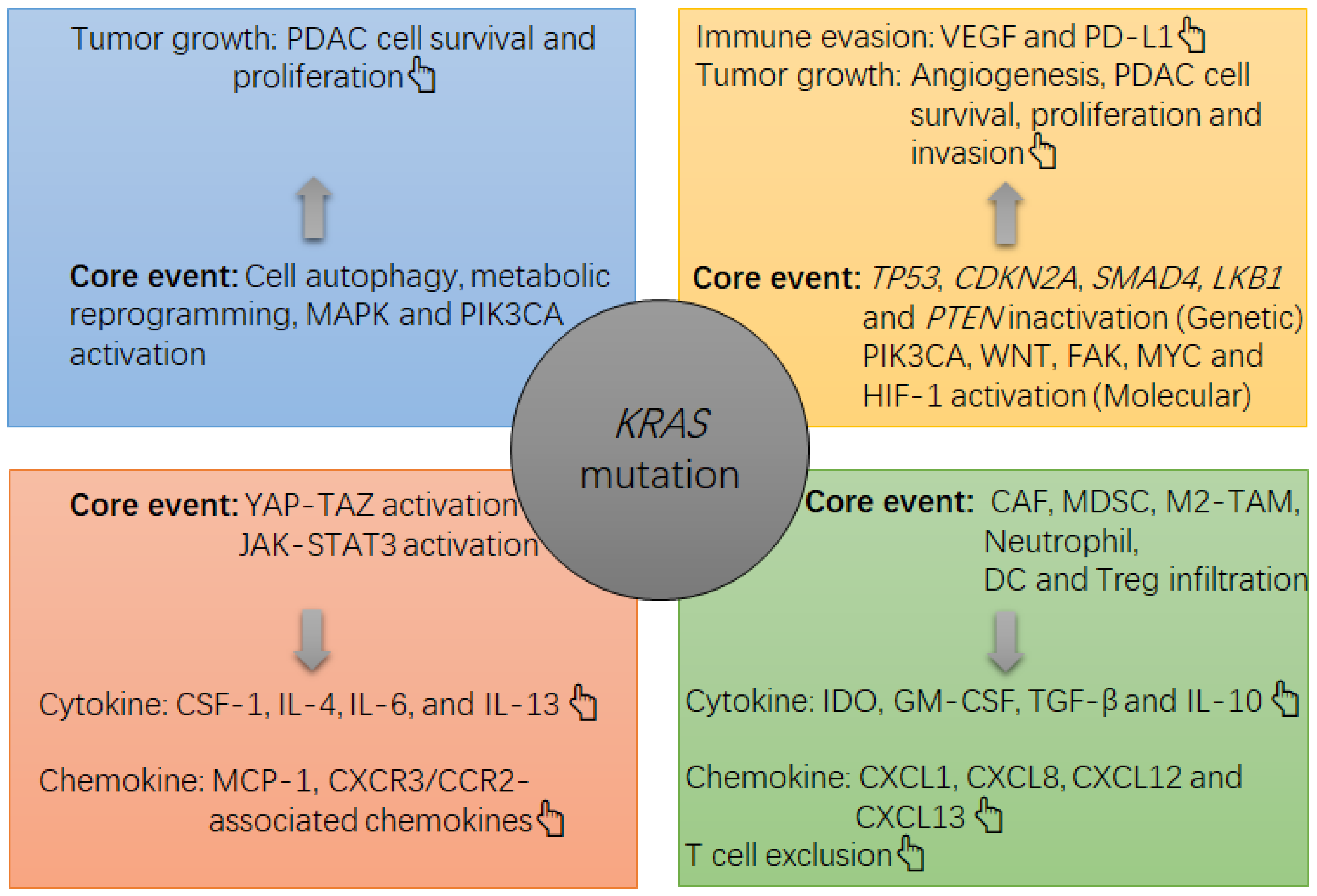
2. The Carcinogenic Role of KRAS Mutation in PDAC
3. The KRAS Mutation and Immune Environment in PDAC
4. Current Status of Immune Checkpoint Blockade Therapy for PDAC
5. Value of KRAS Mutation for Predicting Cancer Immune Status in Other Adenocarcinomas
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Herman, J.; Schulik, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Sohal, D.P.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 27, 3217–3230. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Meng, Q.; Hua, J.; Yu, X.; Shi, S. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: New insights and therapeutic implications. Mol. Cancer. 2019, 18, 184. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Gruber, R.; Panayiotou, R.; Nye, E.; Spencer-Dene, B.; Stamp, G.; Behrens, A. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016, 151, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Humpton, T.J.; Alagesan, B.; deNicola, G.M.; Lu, D.; Yordanov, G.N.; Leonhardt, C.S.; Yao, M.A.; Alagesan, P.; Zaatari, M.N.; Park, Y.; et al. Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov. 2019, 9, 1268–1287. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496. [Google Scholar] [CrossRef]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3021–3024. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Tong, W.; Ma, S.; Chang, P. Standard therapies: Solutions for improving therapeutic effects of immune checkpoint inhibitors on colorectal cancer. OncoImmunology 2020, 9, 1773205. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a0311435. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [Green Version]
- Kerr, E.M.; Gaude, E.; Turrell, F.K.; Frezza, C.; Martins, C.P. Mutant KRAS copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016, 531, 110–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vakiani, E.; Janakiraman, M.; Shen, R.; Sinha, R.; Zeng, Z.; Shia, J.; Cercek, A.; Kemeny, N.; D’Angelica, M.; Viale, A.; et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J. Clin. Oncol. 2012, 30, 2956–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Xu, T.; Chang, P. KRAS/LKB1 and KRAS/TP53 co-mutations create divergent immune signatures in lung adenocarcinomas. Ther. Adv. Med. Oncol. 2020. [Google Scholar] [CrossRef]
- Lal, N.; White, B.S.; Goussous, G.; Pickles, O.; Mason, M.J.; Beggs, A.D.; Taniere, P.; Willcox, B.E.; Guinney, J.; Middleton, G.W. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clin. Cancer Res. 2018, 24, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J., II; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterization of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2020, 70, 148–156. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177: Phase 3, Open-label, randomized study of first-line pembrolizumab (Pembro) versus investigator-choice chemotherapy for mismatch repair-deficient (dMMR) or microsatellite instability-high (MSI-H) metastatic colorectal carcinoma (mCRC). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Gan, L.L.; Hii, L.W.; Wong, S.F.; Leong, C.O.; Mai, C.W. Molecular Mechanisms and Potential Therapeutic Reversal of Pancreatic Cancer-Induced Immune Evasion. Cancers 2020, 12, 1872. [Google Scholar] [CrossRef]
- Sivaram, N.; McLaughlin, P.A.; Han, H.V.; Petrenko, O.; Jiang, Y.P.; Ballou, L.M.; Pham, K.; Liu, C.; van der Velden, A.W.; Lin, R.Z. Tumor-intrinsic PIK3CA represses tumor immunogenecity in a model of pancreatic cancer. J. Clin. Investig. 2019, 129, 3264–3276. [Google Scholar] [CrossRef] [Green Version]
- Boutin, A.T.; Liao, W.T.; Wang, M.; Hwang, S.S.; Karpinets, T.V.; Cheung, H.; Chu, G.C.; Jiang, S.; Hu, J.; Chang, K.; et al. Oncogenic KRAS drives invasion and maintains metastases in colorectal cancer. Genes Dev. 2017, 31, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Muto, G. TGF-β function in immune suppression. Curr. Top Microbiol. Immunol. 2011, 350, 127–147. [Google Scholar]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019, 35, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Wang, J.J.; Yin, P.H.; Xu, K.; Wang, Y.Z.; Shi, F.; Gao, J.; Fu, X.L. Strategies to target energy metabolism in consensus molecular subtype 3 along with Kirsten rat sarcoma viral oncogene homolog mutations for colorectal cancer therapy. J. Cell Physiol. 2019, 234, 5601–5612. [Google Scholar] [CrossRef]
- He, P.; Yang, J.W.; Yang, V.W.; Bialkowska, A.B. Krüppel-like Factor 5, Increased in Pancreatic Ductal Adenocarcinoma, Promotes Proliferation, Acinar-to-Ductal Metaplasia, Pancreatic Intraepithelial Neoplasia, and Tumor Growth in Mice. Gastroenterology 2018, 154, 1494–1508. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Nandakumar, N.; Shi, Y.; Manzano, M.; Smith, A.; Graham, G.; Gupta, S.; Vietsch, E.E.; Laughlin, S.Z.; Wadhwa, M.; et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 2014, 7, ra42. [Google Scholar] [CrossRef] [Green Version]
- De Nicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Chang, C.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Eibl, G.; Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, X.; Fang, L.; Lan, C.; Zheng, X.; Wang, Y.; Zhang, Y.; Han, X.; Liu, S.; Cheng, K.; et al. A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. 2017, 402, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.T.S.; Zeron-Medina, J.; Hernandez, J.; Macarulla, T.; Balsells, J.; Merino, X.; Allende, H.; Tabernero, J. Overexpression of Yes Associated Protein 1, an Independent Prognostic Marker in Patients with Pancreatic Ductal Adenocarcinoma, Correlated with Liver Metastasis and Poor Prognosis. Pancreas 2017, 46, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yang, S.; Zhang, F.; Cheng, F.; Wang, X.; Rao, J. Influence of the Hippo-YAP signaling pathway on tumor associated macrophages (TAMs) and its implications on cancer immunosuppressive microenvironment. Ann. Transl. Med. 2020, 8, 399. [Google Scholar]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KRASG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Shabhazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Li, S.; Xu, H.X.; Wu, C.T.; Wang, W.Q.; Jin, W.; Gao, H.L.; Li, H.; Zhang, S.R.; Xu, J.Z.; Qi, Z.H.; et al. Angiogenesis in pancreatic cancer: Current research status and clinical implications. Angiogenesis 2019, 22, 15–36. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J. Natl. Cancer Inst. 2017, 109, djw283. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.A., III; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergized with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extracuumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Luke, J.J.; Lemons, J.M.; Karrison, T.G.; Pitroda, S.P.; Melotek, J.M.; Zha, Y.; Al-Hallaq, H.A.; Arina, A.; Khodarev, N.N.; Janisch, L.; et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1611–1618. [Google Scholar] [CrossRef]
- Xie, C.; Duffy, A.G.; Brar, G.; Fioravanti, S.; Mabry-Hrones, D.; Walker, M.; Bonilla, C.M.; Wood, B.J.; Citrin, D.E.; Ramirez, E.M.G.; et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ju, X.; Wang, J.; Fan, Y.; Ren, M.; Zhang, H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018, 438, 17–23. [Google Scholar] [CrossRef]
- Weiss, G.J.; Waypa, J.; Blaydorn, L.; Coats, J.; McGahey, K.; Sangal, A.; Niu, J.; Lynch, C.A.; Farley, J.H.; Khemka, V. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PmebroPlus). Br. J. Cancer 2017, 117, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., III; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef] [Green Version]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose, escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; George, B.; Gutierrez, M.; Johns, M.E.; Chiorean, E.G.; Kwak, E.L.; Kalyan, A.; Manax, V.; Ye, M.; et al. Phase I study of nivolumab (nivo) + nab-paclitaxel (nab-P) + gemcitabine (Gem) in advanced pancreatic cancer (APC). J. Clin. Oncol. 2019, 37, 298. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; George, B.; Gutierrez, M.; Johns, M.E.; Chiorean, E.G.; Kwak, E.L.; Kalyan, A.; Manax, V.; Ye, M.; et al. Phase I study of nivolumab (nivo) + nab-paclitaxel (nab-P) ± gemcitabine (Gem) in solid tumors: Interim results from the pancreatic cancer (PC) cohorts. J. Clin. Oncol. 2017, 35, 412. [Google Scholar] [CrossRef]
- Renouf, D.J.; Dhani, N.C.; Kavan, P.; Jonker, D.J.; Wei, A.C.C.; Hsu, T.; Tang, P.A.; Graham, B.; Gallinaro, L.; Hasan, T.; et al. The Canadian Cancer Trials Group PA.7 trial: Results from the safety run in of a randomized phase II study of gemcitabine (GEM) and nab-paclitaxel (Nab-P) versus GEM, nab-P, durvalumab (D), and tremelimumab (T) as first-line therapy in metastatic pancreatic ductal adenocarcinoma (mPDAC). J. Clin. Oncol. 2018, 36, 349. [Google Scholar]
- Borazanci, E.H.; Jameson, G.S.; Borad, M.J.; Ramanathan, R.K.; Korn, R.L.; Caldwell, L.; Ansaldo, K.; Hendrickson, K.; Marceau, K.; von Hoff, D.D. A phase II pilot trial of nivolumab (N) + albumin bound paclitaxel (AP) + paricalcitol (P) + cisplatin (C) + gemcitabine (G) (NAPPCG) in patients with previously untreated metastatic pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2018, 36, 358. [Google Scholar] [CrossRef]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1107. [Google Scholar] [PubMed]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Green, M.D.; Lang, X.; Lazarus, J.; Parsels, J.D.; Wei, S. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res. 2019, 79, 3940–3951. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.A.; Pilié, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef] [Green Version]
- Keenan, B.P.; Saenger, Y.; Kafrouni, M.I.; Leubner, A.; Lauer, P.; Maitra, A.; Rucki, A.A.; Gunderson, A.J.; Coussens, L.M.; Brockstedt, D.G.; et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology 2014, 146, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Blair, A.B.; Kleponis, J.; Thomas, D.L.; Muth, S.T.; Murphy, A.G.; Kim, V.; Zheng, L. IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma. J. Clin. Investig. 2019, 129, 1742–1755. [Google Scholar] [CrossRef]
- Burrack, A.L.; Spartz, E.J.; Raynor, J.F.; Wang, I.; Olson, M.; Stromnes, I.M. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep. 2019, 28, 2140–2155. [Google Scholar] [CrossRef] [Green Version]
- Christenson, E.S.; Jaffee, E.; Azad, N.S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol. 2020, 21, e135–e145. [Google Scholar] [CrossRef]
- Nollmann, F.I.; Ruess, D.A. Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising? Biomedicines 2020, 8, 281. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Briere, D.; Calinisan, A.; Aranda, R.; Sudhakar, N.; Hargis, L.; Gatto, S. The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Leads to Durable Complete Responses in Combination with Anti-PD-1 Therapy in a Syngeneic Mouse Model. Mol. Cancer Ther. 2021. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Smeby, J.; Sveen, A.; Merok, M.A.; Danielsen, S.A.; Eilertsen, I.A.; Guren, M.G.; Dienstmann, R.; Nesbakken, A.; Lothe, R.A. CMS-dependent prognostic impact of KRAS and BRAFV600E mutations in primary colorectal cancer. Ann. Oncol. 2018, 29, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Kim, T.W.; van Cutsem, E.; Geva, R.; Jager, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Yuki, S.; Bando, H.; Tsukada, Y.; Inamori, K.; Komatsu, Y.; Homma, S.; Uemura, M.; Kato, T.; Kotani, D.; Fukuoka, S.; et al. Short-term results of VOLTAGE-A: Nivolumab monotherapy and subsequent radical surgery following preoperative chemoradiotherapy in patients with microsatellite stable and microsatellite instability-high locally advanced rectal cancer. J. Clin. Oncol. 2020, 38, 4100. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Chibaudel, B.; Taieb, J.; Bennouna, J.; Martin-Babau, J.; Fonck, M.; Borg, C.; Cohen, R.; Thibaudin, M.; Limagne, E.; et al. Durvalumab and tremelimumab in combination with FOLFOX in patients with RAS-mutated, microsatellite-stable, previously untreated metastatic colorectal cancer (mCRC): Results of the first intermediate analysis of the phase Ib/II MEDETREME trial. J. Clin. Oncol. 2020, 38, 3006. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients with Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef]

{kind=link}
| Cancer | PDAC | CRAC | LUAC | |
|---|---|---|---|---|
| Characters [Ref.] | ||||
| Prevalence of KRAS mutation | 97.7% [9] | 44.7% [9] | 30.9% [9] | |
| Hottest missense mutation in KRAS | G12D [9] | G12D [9] | G12C [9] | |
| Sensitive to glucose restriction vs. KRASwt | Yes [19] | Yes [20] | No [21] | |
| Common alteration with KRAS | TP53 inactivation [10] | TP53 and APC inactivation [22] | TP53 or LKB1 inactivation [23] | |
| General milieu of KRAS-mutant tumors | Immune-cold [7] | Immune-cold [24] | KRAS-only: immune-cold or hot [23] TP53 inactivation: immune-hot [23] LKB1 inactivation: immune-cold [23] | |
| Number/function of tumoricidal T cells in KRAS-mutant tumors | Decrease/Decrease [7] | Decrease/Decrease [24] | KRAS-only: slight increase/decrease [23] TP53 inactivation: significant increase/decrease [23] LKB1 inactivation: significant decrease/decrease [23] | |
| Major type of immune infiltrates in KRAS-mutant tumors | Myeloid suppressive cell [7] | Myeloid suppressive cell [24] | KRAS-only: T cell, macrophage, neutrophil [23] TP53 inactivation: CD8+ T cell, CD45RO+ T cell [23] LKB1 inactivation: myeloid suppressive cell [23] | |
| Common presentation of the ICB therapy biomarker if KRAS mutation | pMMR/MSS [25] | pMMR/MSS [26] | KRAS-only: PD-L1 expression ↑ [23] TP53 inactivation: PD-L1 expression ↑↑ [23] LKB1 inactivation: PD-L1 expression ↓↓ [23] | |
| Biomarker associated with the effectiveness of ICB therapy | dMMR/MSI-H [4] | dMMR/MSI-H [27] | PD-L1 [23] | |
| Prevalence of dMMR/MSI-H in all cases | 1~2% [25] | 14% [26] | NM | |
| Prevalence of positive expression of PD-L1 by tumor cells | NM | NM | Among KRAS-only tumors: 37.5% [23] Among TP53 inactivation tumors: 68.8% [23] Among LKB1 inactivation tumors: 10% [23] | |
| General response to monotherapy using ICB drugs | Poor [5] | Poor [28] | KRAS-only tumor: Fair [23] TP53 inactivation tumor: Excellent [23] LKB1 inactivation tumor: Poor [23] | |
| Core molecular events associated with KRAS mutation-induced immunosuppression | 1. YAP-TAZ activation [12]; 2. JAK-STAT3 activation [12]; 3. Metabolic reprogramming of glucose and cell autophagy [13,14]; 4. In concert with other events, TP53 inactivation [15], LKB1 mutation [29,30], PTEN loss [29,30], WNT/β-catenin activation [29,30], FAK activation [29,30], PIK3CA activation [29,30] and MYC activation [29,30]; | 1. In concert with APC and TP53 inactivation: TGF-β1 upregulation and EMT [31]; 2. TGF-β-induced immune suppression [32]; 3. IRF2 inactivation [24,33]; 4. Metabolic dysregulation in glucose, glutamine, fatty acid and lipid [26,34]; 5. MAPK and HIF-1-related cascade activation [34]; | 1. ERK activation-induced PD-L1 upregulation [23] 2. Metabolic reprogramming of glucose [21] 3. In concert with LKB1 inactivation: strengthening metabolic reprogramming of glucose and JAK-STAT3 activation [23] | |
| Author [Ref.] | Year | Phase | Patient No. | ICB Drug | Other Treatment | ORR |
|---|---|---|---|---|---|---|
| • First-line therapy | ||||||
| Aglietta M, et al. [62] | 2014 | I | 34 | Tremelimumab | Gemcitabine | 10.5% |
| Wainberg ZA, et al. [63] | 2019 | I | 50 | Nivolumab | Gemcitabine + Nab- paclitaxel | 18% |
| Wainberg ZA, et al. [64] | 2017 | I | 17 | Nivolumab | Gemcitabine + Nab- paclitaxel | 50% |
| Renouf, et al. [65] | 2018 | II | 11 | Durvalumab + Tremelimumab | Gemcitabine + Nab-paclitaxel | 73% |
| Borazanci, et al. [66] | 2018 | II | 11 | Nivolumab | Gemcitabine + Nab-paclitaxel + Cisplatin + Paricalcitol | 80% |
| • Second- or later-line therapy | ||||||
| Luke JJ, et al. [57] | 2018 | I | 3 | Pembrolizumab | SBRT: 30–50 Gy for 2–4 metastatic lesions | NR |
| O’Reilly EM, et al. [56] | 2019 | II | Arm A: 32 Arm B: 32 | Durvalumab Durvalumab + Tremelimumab | No | 0% 3.1% |
| Xie C, et al. [58] | 2020 | I | Arm A1: 14 Arm A2: 10 Arm B1: 19 Arm B2: 16 | Durvalumab Durvalumab Durvalumab + Tremelimumab Durvalumab + Tremelimumab | SBRT: 8 Gy/1 fraction SBRT: 25 Gy/5 fractions SBRT: 8 Gy/1 fraction SBRT: 25 Gy/5 fractions | 5.1% A |
| Weiss GJ, et al. [60] | 2017 | I | 11 | Pembrolizumab | Gemcitabine (Gem)-based chemotherapy | 18.2% |
| Kamath SD, et al. [61] | 2020 | I | 21 B | Arm A: Ipilimumab 3 mg/kg Arm B: Ipilimumab 3 mg/kg Arm C: Ipilimumab 6 mg/kg | Gem 750 mg/m2 Gem 1g/m2 Gem 1g/m2 | 14% C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, M.; Gao, Y.; Chang, P. KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers 2021, 13, 2429. https://doi.org/10.3390/cancers13102429
Gu M, Gao Y, Chang P. KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers. 2021; 13(10):2429. https://doi.org/10.3390/cancers13102429
Chicago/Turabian StyleGu, Meichen, Yanli Gao, and Pengyu Chang. 2021. "KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas" Cancers 13, no. 10: 2429. https://doi.org/10.3390/cancers13102429
APA StyleGu, M., Gao, Y., & Chang, P. (2021). KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers, 13(10), 2429. https://doi.org/10.3390/cancers13102429