
Who’s Driving? Switch of Drivers in Immunotherapy-Treated Progressing Sinonasal Melanoma

 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
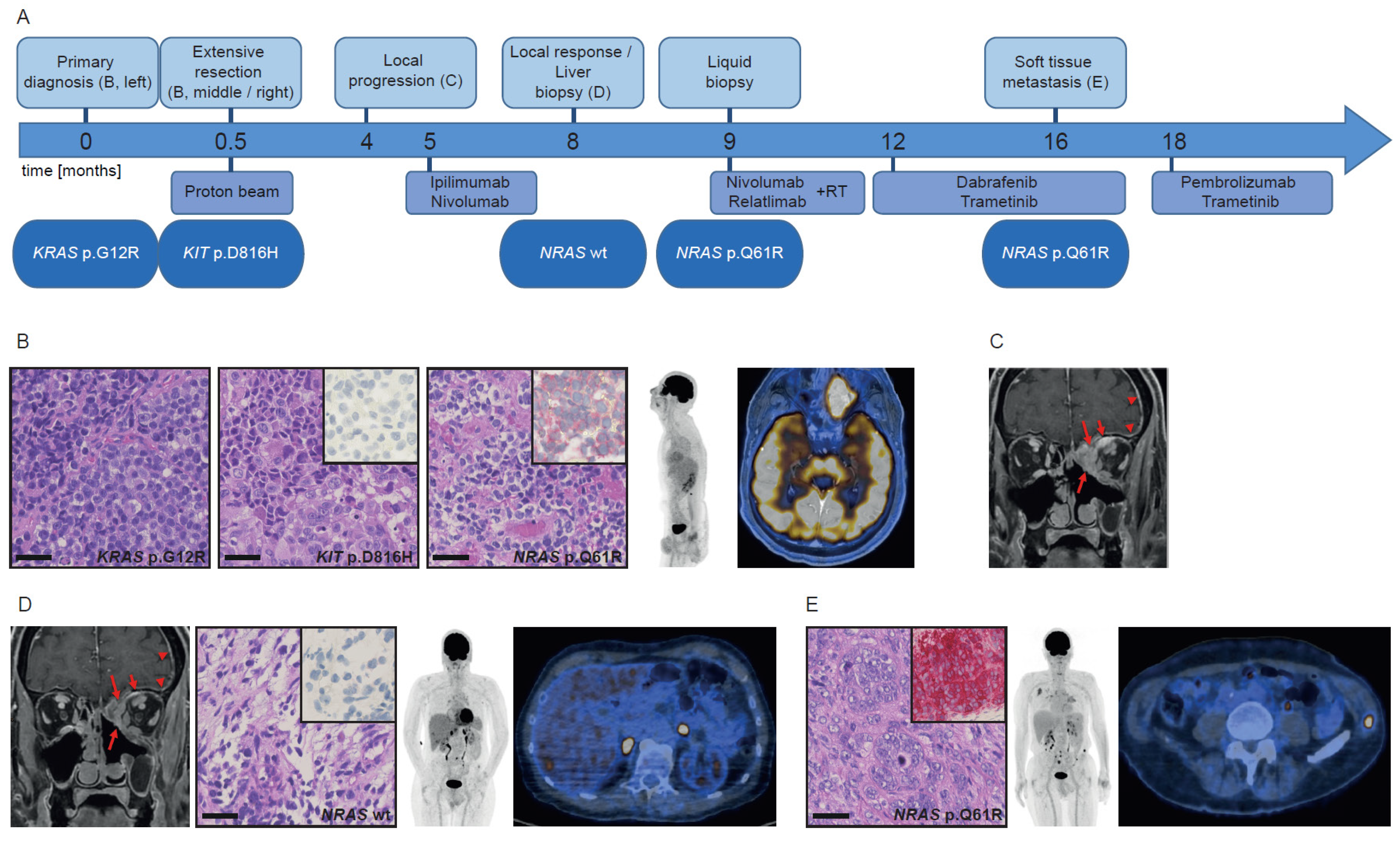
3.1. Patient History Shows Emergence and Selection of an NRAS-Mutant Clone during the Course of Immunotherapy
3.1.1. Case 1
3.1.2. Case 2
3.1.3. Case 3
3.2. Mutational Analysis of Different Tumors Reveals Both Intra-Patient Similarities and Molecular Differences
3.3. NRAS Clones either Preexisted or Emerged during the Course of Disease and Therapy
3.4. Tumor Morphology Revealed No Association with Mutational Status or Response to Therapy
3.5. CD8+ T Cell Infiltration, PD-L1 Expression and Tumor Mutational Burden (TMB) Do Not Correlate with Therapy Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ascierto, P.A.; Accorona, R.; Botti, G.; Farina, D.; Fossati, P.; Gatta, G.; Gogas, H.; Lombardi, D.; Maroldi, R.; Nicolai, P.; et al. Mucosal melanoma of the head and neck. Crit. Rev. Oncol. Hematol. 2017, 112, 136–152. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (review). Int. J. Oncol. 2018, 52, 1071. [Google Scholar] [CrossRef] [Green Version]
- Freiberger, S.N.; Morand, G.B.; Turko, P.; Wager, U.; Dummer, R.; Hüllner, M.; Holzmann, D.; Rupp, N.J.; Levesque, M.P. Morpho-molecular assessment indicates new prognostic aspects and personalized therapeutic options in sinonasal melanoma. Cancers 2019, 11, 1329. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Keilholz, U. Appendix 2: Cutaneous Melanoma (2): EUpdate Published Online September 2016. Ann. Oncol. 2016, 27, v136–v137. [Google Scholar] [CrossRef]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified kit arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebbe, C.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.-P.; Avril, M.-F.; Bedane, C.; Bens, G.; et al. Phase II multicentric uncontrolled national trial assessing the efficacy of nilotinib in the treatment of advanced melanomas with c-kit mutation or amplification. JCO 2014, 32, 9032. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819. [Google Scholar] [CrossRef]
- Wagle, N.; Allen, E.M.V.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Loo, P.V.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751. [Google Scholar] [CrossRef]
- Hao, J.-J.; Lin, D.-C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.-Y.; Chang, C.; Jiang, Y.; Lu, C.-C.; Shi, Z.-Z.; Xu, X.; et al. Spatial intratumor heterogeneity of genetic, epigenetic alterations and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500. [Google Scholar] [CrossRef]
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-Existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, C.; Cardelli, L.; Padova, M.; Nardo, L.; Ciciarelli, V.; Rocco, T.; Cipolloni, G.; Clementi, M.; Cortellini, A.; Ventura, A.; et al. Intra-patient heterogeneity of BRAF and NRAS molecular alterations in primary melanoma and metastases. Acta Derm. Venerol. 2020, 100, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchberger, M.C.; Ugurel, S.; Mangana, J.; Heppt, M.V.; Eigentler, T.K.; Berking, C.; Schadendorf, D.; Schuler, G.; Dummer, R.; Heinzerling, L. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 2018, 98, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Chatila, W.K.; Arora, A.; Sanchez-Vega, F.; Kantheti, H.S.; Rojas Zamalloa, J.A.; Krieger, P.; Callahan, M.K.; Betof Warner, A.; Postow, M.A.; et al. Therapeutic implications of detecting MAPK-activating alterations in cutaneous and unknown primary melanomas. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Johnson, D.B.; Lovly, C.M.; Flavin, M.; Panageas, K.S.; Ayers, G.D.; Zhao, Z.; Iams, W.T.; Colgan, M.; DeNoble, S.; Terry, C.R.; et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol. Res. 2015, 3, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Moya-Plana, A.; Herrera Gómez, R.G.; Rossoni, C.; Dercle, L.; Ammari, S.; Girault, I.; Roy, S.; Scoazec, J.-Y.; Vagner, S.; Janot, F.; et al. Evaluation of the efficacy of immunotherapy for non-resectable mucosal melanoma. Cancer Immunol. Immunother. 2019, 68, 1171–1178. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Larkin, J.; Sosman, J.A.; Lebbé, C.; Brady, B.; Neyns, B.; Schmidt, H.; Hassel, J.C.; Hodi, F.S.; Lorigan, P.; et al. Efficacy and safety of nivolumab alone or in combination with ipilimumab in patients with mucosal melanoma: A pooled analysis. JCO 2016, 35, 226–235. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–pd-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierauf, J.; Veit, J.A.; Affolter, A.; Bergmann, C.; Grünow, J.; Laban, S.; Lennerz, J.K.; Grünmüller, L.; Mauch, C.; Plinkert, P.K.; et al. Identification and clinical relevance of pd-l1 expression in primary mucosal malignant melanoma of the head and neck. Melanoma Res. 2015, 25, 503–509. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to t cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.; Kakavand, H.; Alexandrov, L.B.; Mann, G.J.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Mutations | Frequency | Molecular Depth | Tumor Cell Content | Comment | |
|---|---|---|---|---|---|
| Patient 1 | KRAS p.G12R NRAS p.Q61R NRAS p.G12V | 4.6% 3.27% 7.04% | 913x 765x 270x | 40% | KIT not covered by the assay |
| Patient 2 | KRAS p.G12A | 64.21% | 992x | 70% | |
| Patient 3 | WT | 30% |
| Size | Morphology | Nuclei | Other Features | Main Molecular Alteration | |
|---|---|---|---|---|---|
| Patient 1 | |||||
| Primary diagnosis | medium/large | pleomorphic, atypical | variable size, prominent nucleoli, eccentric nuclei | eosinophilic cytoplasm, abundant mitoses | KRAS p.G12R |
| Extensive resection | medium/large | pleomorphic, atypical | convoluted nuclei, variable size, prominent nucleoli, eccentric nuclei | eosinophilic cytoplasm, abundant mitoses | KIT p.D816H |
| Liver biopsy | few vital cells, myxoid stromal changes | WT | |||
| Soft tissue metastasis | medium/large | pleomorphic, atypical | chromatin clearing of the nuclei | eosinophilic cytoplasm, abundant mitoses | NRAS p.Q61R |
| Patient 2 | |||||
| Primary diagnosis | medium/large | monomorphic, atypical | variously distinct nucleoli | eosinophilic cytoplasm | KRAS p.G12A/amplification |
| Local recurrence 1 | spindle cells: medium/large; epithelioid cells: small | biphasic appearance (spindle cell shaped, monomorphic epithelioid) | NRAS p.Q61K | ||
| Local recurrence 2 | small/ medium | monomorphic, atypical | NRAS p.Q61K | ||
| Patient 3 | |||||
| Primary diagnosis | medium/large | atypical, epithelioid | prominent nucleoli | eosinophilic cytoplasm, abundant mitoses | WT |
| LN level II | medium/large | atypical, epithelioid | prominent nucleoli | eosinophilic cytoplasm, abundant mitoses | WT |
| Hemi- colectomy | medium/large | atypical, epithelioid | prominent nucleoli | eosinophilic cytoplasm, abundant mitoses | PIK3CA p.C420R |
| Adrenal gland metastasis | small/ medium | atypical, epithelioid | lacking prominent nucleoli | eosinophilic cytoplasm, abundant mitoses | NRAS p.Q61K |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freiberger, S.N.; Turko, P.; Hüllner, M.; Dummer, R.; Morand, G.B.; Levesque, M.P.; Holzmann, D.; Rupp, N.J. Who’s Driving? Switch of Drivers in Immunotherapy-Treated Progressing Sinonasal Melanoma. Cancers 2021, 13, 2725. https://doi.org/10.3390/cancers13112725
Freiberger SN, Turko P, Hüllner M, Dummer R, Morand GB, Levesque MP, Holzmann D, Rupp NJ. Who’s Driving? Switch of Drivers in Immunotherapy-Treated Progressing Sinonasal Melanoma. Cancers. 2021; 13(11):2725. https://doi.org/10.3390/cancers13112725
Chicago/Turabian StyleFreiberger, Sandra N., Patrick Turko, Martin Hüllner, Reinhard Dummer, Grégoire B. Morand, Mitchell P. Levesque, David Holzmann, and Niels J. Rupp. 2021. "Who’s Driving? Switch of Drivers in Immunotherapy-Treated Progressing Sinonasal Melanoma" Cancers 13, no. 11: 2725. https://doi.org/10.3390/cancers13112725