
Biomarkers for Diagnosis, Prognosis and Response to Immunotherapy in Melanoma

 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathogenic and Epidemiological Diagnostic Biomarkers in Melanoma
2.1. Classic Diagnostic Criteria and Markers
2.2. Tumor Specific Lymphatic Vessel Biomarkers
2.3. Genetic Diagnostic Markers
2.4. Lymph Node Evaluation
2.5. New Possible Diagnostic Parameters
3. Prognostic and Predictive Biomarkers
3.1. Genetic Prognostic Factors
3.2. Prognostic Factors in Lymphangiogenesis
3.3. Lymph Node Prognostic Role
3.4. Prognostic and Predictive Biomarkers and Immunotherapy
3.5. miRNAs as Possible Biomarkers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer International Publishing: New York, NY, USA, 2017. [Google Scholar]
- Tracey, E.H.; Vij, A. Updates in Melanoma. Dermatol. Clin. 2019, 37, 73–82. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A.; Hess, K.R.; Sondak, V.K.; Long, G.V.; Ross, M.I.; Lazar, A.J.; Faries, M.B.; Kirkwood, J.M.; McArthur, G.A.; et al. Melanoma staging: Evidence-based changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 472–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, D., III; Aung, P.P.; Jour, G. The “-OMICS” facet of melanoma: Heterogeneity of genomic, proteomic and metabolomic biomarkers. Semin. Cancer Biol. 2019, 59, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Targeting invasive properties of melanoma cells. FEBS J. 2017, 284, 2148–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.R.; Luo, L.; Berwick, M. Sex Differences in Melanoma. Curr. Epidemiol. Rep. 2019, 6, 112–118. [Google Scholar] [CrossRef]
- Courtenay, W. Behavioral Factors Associated with Disease, Injury, and Death among Men: Evidence and Implications for Prevention. J. Men Stud. 2000, 9, 81–142. [Google Scholar] [CrossRef]
- Paddock, L.E.; Lu, S.E.; Bandera, E.V.; Rhoads, G.G.; Fine, J.; Paine, S.; Barnhill, R.; Berwick, M. Skin self-examination and long-term melanoma survival. Melanoma Res. 2016, 26, 401–408. [Google Scholar] [CrossRef]
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.; Hölzel, D.; Coebergh, J.W.W.; Engel, J. Gender Differences in Melanoma Survival: Female Patients Have a Decreased Risk of Metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Artomov, M.; Goggins, W.; Daly, M.; Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. J. Natl. Cancer Inst. 2015, 107, djv211. [Google Scholar] [CrossRef]
- Nonaka, D.; Chiriboga, L.; Rubin, B.P. Differential expression of S100 protein subtypes in malignant melanoma, and benign and malignant peripheral nerve sheath tumors. J. Cutan. Pathol. 2008, 35, 1014–1019. [Google Scholar] [CrossRef]
- Ribé, A.; McNutt, N.S.; Rib, A. S100A6 Protein Expression is Different in Spitz Nevi and Melanomas. Mod. Pathol. 2003, 16, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Eisenstein, A.; Gonzalez, E.C.; Raghunathan, R.; Xu, X.; Wu, M.; McLean, E.O.; McGee, J.; Ryu, B.; Alani, R.M. Emerging Biomarkers in Cutaneous Melanoma. Mol. Diagn. Ther. 2018, 22, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, D.; Leininger, J.; Hamby, C.; Safai, B. Diagnostic and Prognostic Biomarkers in Melanoma. J. Clin. Aesthet. Dermatol. 2014, 7, 13–24. [Google Scholar] [PubMed]
- Ordóñez, N.G. Value of melanocytic associated immunohistochemical markers in the diagnosis of malignant melanoma: A review and update. Hum. Pathol. 2014, 45, 191–205. [Google Scholar] [CrossRef]
- Salazar-Onfray, F.; López, M.; Lundqvist, A.; Aguirre, A.; Escobar, A.; Serrano, A.; Korenblit, C.; Petersson, M.; Chhajlani, V.; Larsson, O.; et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br. J. Cancer 2002, 87, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blochin, E.; Nonaka, D. Diagnostic value of Sox10 immunohistochemical staining for the detection of metastatic melanoma in sentinel lymph nodes. Histopathology 2009, 55, 626–628. [Google Scholar] [CrossRef]
- Abbas, O.; Miller, D.D.; Bhawan, J. Cutaneous malignant melanoma: Update on diagnostic and prognostic biomarkers. Am. J. Dermatopathol. 2014, 36, 363–379. [Google Scholar] [CrossRef]
- Xu, X.; Gimotty, P.A.; Guerry, D.; Karakousis, G.; Elder, D.E. Lymphatic invasion as a prognostic biomarker in primary cutaneous melanoma. Methods Mol. Biol. 2014, 1102, 275–286. [Google Scholar] [PubMed] [Green Version]
- Špirić, Z.; Erić, M.; Eri, Ž. Lymphatic invasion and the Shields index in predicting melanoma metastases. J. Plast. Reconstr. Aesthet. Surg. 2017, 70, 1646–1652. [Google Scholar] [CrossRef]
- Banerji, S.; Ni, J.; Wang, S.-X.; Clasper, S.; Su, J.; Tammi, R.; Jones, M.; Jackson, D.G. LYVE-1, a New Homologue of the CD44 Glycoprotein, Is a Lymph-specific Receptor for Hyaluronan. J. Cell Biol. 1999, 144, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Kahn, H.J.; Marks, A. A New Monoclonal Antibody, D2-40, for Detection of Lymphatic Invasion in Primary Tumors. Lab. Investig. 2002, 82, 1255–1257. [Google Scholar] [CrossRef] [Green Version]
- Špirić, Z.; Vještica, M.; Erić, M. Survival prediction in patients with cutaneous melanoma by tumour lymphangiogenesis. Acta Clin. Belg. 2020, 75, 379–387. [Google Scholar] [CrossRef]
- Bradish, J.R.; Cheng, L. Molecular pathology of malignant melanoma: Changing the clinical practice paradigm toward a personalized approach. Human Pathol. 2014, 45, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Hugdahl, E.; Kalvenes, M.B.; Puntervoll, H.E.; Ladstein, R.G.; Akslen, L.A. BRAF-V600E ex-pression in primary nodular melanoma is associated with aggressive tumour features and re-duced survival. Br. J. Cancer 2016, 114, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Tsao, H.; Bevona, C.; Goggins, W.; Quinn, T. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: A population-based estimate. Arch. Dermatol. 2003, 139, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psaty, E.L.; Scope, A.; Halpern, A.C.; Marghoob, A.A. Defining the patient at high risk for melanoma. Int. J. Dermatol. 2010, 49, 362–376. [Google Scholar] [CrossRef]
- Gumaste, P.V.; Penn, L.A.; Cymerman, R.M.; Kirchhoff, T.; Polsky, D.; McLellan, B. Skin cancer risk in BRCA1/2 mutation carriers. Br. J. Dermatol. 2015, 172, 1498–1506. [Google Scholar] [CrossRef] [Green Version]
- Garibyan, L.; Fisher, D.E. How Sunlight Causes Melanoma. Curr. Oncol. Rep. 2010, 12, 319–326. [Google Scholar] [CrossRef]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Guterres, A.; Herlyn, M.; Villanueva, J. Melanoma. In eLs; John Wiley and Sons: Hoboken, NJ, USA, 2018; pp. 1–10. [Google Scholar]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., III; Azar, S.; et al. KIT gene mutations and copy number in melanoma sub-types. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef] [Green Version]
- Lange, J.R. Review of the Guidelines on the Management of the Regional Lymph Nodes in Patients with Melanoma. JAMA Surg. 2020, 155, 258. [Google Scholar] [CrossRef]
- Foster, J.M.; Oumie, A.; Togneri, F.S.; Vasques, F.R.; Hau, D.; Taylor, M.; Tinkler-Hundal, E.; Southward, K.; Medlow, P.; McGreeghan-Crosby, K.; et al. Cross-laboratory validation of the OncoScan® FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med. Genom. 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, B.C.; Olshen, A.B.; LeBoit, P.E.; Pinkel, D. Classifying Melanocytic Tumors Based on DNA Copy Number Changes. Am. J. Pathol. 2003, 163, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Kitano, S.; Nakayama, T.; Yamashita, M. Biomarkers for Immune Checkpoint Inhibitors in Melanoma. Front. Oncol. 2018, 8, 270. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemu-rafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1460. [Google Scholar] [CrossRef]
- Ugurel, J.; Rohmel, P.A.; Ascierto, K.T.; Flaherty, J.J.; Grob, A.; Hauschild, J.; Larkin, G.V.; Long, P.; Lorigan, G.A.; McArthur, A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies. Eur. J. Cancer 2016, 53, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Hidaka, T.; Kambayashi, Y.; Aiba, S. BRAF kinase inhibitors for treatment of melanoma: Developments from early-stage animal studies to Phase II clinical trials. Expert Opin. Investig. Drugs 2018, 28, 143–148. [Google Scholar] [CrossRef]
- Sarkisian, S.; Davar, D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des. Dev. Ther. 2018, 12, 2553–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, E.; Zaremba, A.; Horn, S.; Ugurel, S.; Casalini, B.; Schlaak, M.; Hassel, J.C.; Herbst, R.; Utikal, J.S.; Weide, B.; et al. GNAQ and GNA11 mutant nonuveal melanoma: A sub-type distinct from both cutaneous and uveal melanoma. Br. J. Dermatol. 2020, 183, 928–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viros, A.; Sanchez-Laorden, B.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 2014, 511, 478–482. [Google Scholar] [CrossRef]
- Zebary, A.; Omholt, K.; van Doorn, R.; Ghiorzo, P.; Harbst, K.; Johansson, C.H.; Höiom, V.; Jönsson, G.; Pjanova, D.; Puig, S.; et al. Somatic BRAF and NRAS mutations in familial melanomas with known germline CDKN2A status: A GenoMEL study. J. Investig. Dermatol. 2014, 134, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Pasquali, S.; van der Ploeg, A.P.; Mocellin, S.; Stretch, J.R.; Thompson, J.F.; Scolyer, R.A. Lymphatic biomarkers in primary melanomas as predictors of regional lymph node metastasis and patient outcomes. Pigment Cell Melanoma Res. 2013, 26, 326–337. [Google Scholar] [CrossRef]
- Christianson, D.R.; Dobroff, A.S.; Proneth, B.; Zurita, A.J.; Salameh, A.; Dondossola, E.; Makino, J.; Bologa, C.G.; Smith, T.L.; Makino, J.; et al. Ligand-directed targeting of lymphatic vessels uncovers mechanistic insights in melanoma metastasis. Proc. Natl. Acad. Sci. USA 2015, 112, 2521–2526. [Google Scholar] [CrossRef] [Green Version]
- Broggi, M.A.; Maillat, L.; Clement, C.C.; Bordry, N.; Corthésy, P.; Auger, A.; Matter, M.; Hamelin, R.; Potin, L.; Demurtas, D.; et al. Tumor-associated factors are enriched in lymphatic exudate compared to plasma in metastatic melanoma patients. J. Exp. Med. 2019, 216, 1091–1107. [Google Scholar] [CrossRef] [Green Version]
- García-Silva, S.; Benito-Martín, A.; Sánchez-Redondo, S.; Hernández-Barranco, A.; Ximénez-Embún, P.; Nogués, L.; Mazariegos, M.S.; Brinkmann, K.; López, A.A.; Meyer, L.; et al. Use of extracellular vesicles from lymphatic drainage as surrogate markers of melanoma progression and BRAFV600E mutation. J. Exp. Med. 2019, 216, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Gershenwald, J.E.; Thompson, W.; Mansfield, P.F.; Lee, J.E.; Colome, M.I.; Tseng, C.H.; Lee, J.J.; Balch, C.M.; Reintgen, D.S.; Ross, M.I. Multi-institutional melanoma lymphatic mapping experience: The prognostic value of sentinel lymph node status in 612 stage I or II melanoma patients. J. Clin. Oncol. 1999, 17, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, V.C.; Kirkwood, J.M. State of melanoma: An historic overview of a field in transition. Hematol. Oncol. Clin. N. Am. 2014, 28, 415–435. [Google Scholar] [CrossRef] [Green Version]
- Faries, M.B.; Thompson, J.F.; Cochran, A.J.; Andtbacka, R.H.; Mozzillo, N.; Zager, J.S.; Jahkola, T.; Bowles, T.L.; Testori, A.; Beitsch, P.D.; et al. Completion Dissection or Observation for Sentinel-Node Metastasis in Melanoma. N. Engl. J. Med. 2017, 376, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Leiter, U.; Stadler, R.; Mauch, C.; Hohenberger, W.; Brockmeyer, N.; Berking, C.; Sunderkötter, C.; Kaatz, M.; Schulte, K.W.; Lehmann, P.; et al. Complete lymph node dissection versus no dissection in patients with sentinel lymph node biopsy positive melanoma (DeCOG-SLT): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2016, 17, 757–767. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Borgognoni, L.; Botti, G.; Guida, M.; Marchetti, P.; Mocellin, S.; Muto, P.; Palmieri, G.; Patuzzo, R.; Quaglino, P.; et al. New paradigm for stage III melanoma: From surgery to adjuvant treatment. J. Transl. Med. 2019, 17, 266. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Havel, J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Ouyang, Z.; Wu, H.; Li, L.; Luo, Y.; Li, X.; Huang, G. Regulatory T cells in the immunotherapy of melanoma. Tumor Biol. 2016, 37, 77–85. [Google Scholar] [CrossRef]
- Tietze, J.K.; Angelova, D.; Heppt, M.V.; Reinholz, M.; Murphy, W.J.; Spannagl, M.; Ruzicka, T.; Berking, C. The proportion of circulating CD45RO+ CD8+ memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur. J. Cancer 2017, 75, 268–279. [Google Scholar] [CrossRef]
- Nonomura, Y.; Otsuka, A.; Nakashima, C.; Seidel, J.; Kitoh, A.; Dainichi, T.; Nakajima, S.; Sawada, Y.; Matsushita, S.; Aoki, M.; et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. OncoImmunology 2016, 5, e1248327. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kiyohara, Y.; Uhara, H.; Iizuka, H.; Uehara, J.; Otsuka, F.; Fujisawa, Y.; Takenouchi, T.; Isei, T.; Iwatsuki, K.; et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci. 2017, 108, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- Morello, S.; Capone, M.; Sorrentino, C.; Giannarelli, D.; Madonna, G.; Mallardo, D.; Grimaldi, A.M.; Pinto, A.; Ascierto, P.A. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J. Transl. Med. 2017, 15, 1–9. [Google Scholar] [CrossRef]
- Hutarew, G. PD-L1 testing, fit for routine evaluation? From a pathologist’s point of view. Memo Mag. Eur. Med. Oncol. 2016, 9, 201–206. [Google Scholar]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nat. Cell Biol. 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Yusko, E.; Vignali, M.; Wilson, R.K.; Mardis, E.R.; Hodi, F.S.; Horak, C.; Chang, H.; Woods, D.M.; Robins, H.; Weber, J. Association of Tumor Microenvironment T-cell Repertoire and Mutational Load with Clinical Outcome after Sequential Checkpoint Blockade in Melanoma. Cancer Immunol. Res. 2019, 7, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and an-ti-PD-1 check-point blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Fankhauser, M.; Broggi, M.A.S.; Potin, L.; Bordry, N.; Jeanbart, L.; Lund, A.W.; Da Costa, E.; Hauert, S.; Rincon-Restrepo, M.; Tremblay, C.; et al. Tumor lymphangiogenesis promotes T cell infiltration and potentiates immunotherapy in melanoma. Sci. Transl. Med. 2017, 9, eaal4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Špirić, Z.; Eri, Ž.; Erić, M. Lymphatic vessel density and VEGF-C expression as independent predictors of melanoma metastases. J. Plast. Reconstr. Aesthet. Surg. 2017, 70, 1653–1659. [Google Scholar] [CrossRef]
- Moreira, A.; Leisgang, W.; Schuler, G.; Heinzerling, L. Eosinophilic count as a biomarker for prognosis of melanoma patients and its importance in the response to immunotherapy. Immunotherapy 2017, 9, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.-H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [Green Version]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 36, 740–744. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pontecorvi, G.; Bellenghi, M.; Puglisi, R.; Carè, A.; Mattia, G. Tumor-derived extracellular vesicles and microRNAs: Functional roles, diagnostic, prognostic and therapeutic options. Cytokine Growth Factor Rev. 2020, 51, 75–83. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leidinger, P.; Keller, A.; Borries, A.; Reichrath, J.; Rass, K.; Jager, S.U.; Lenhof, H.-P.; Meese, E. High-throughput miRNA profiling of human melanoma blood samples. BMC Cancer 2010, 10, 262. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, R.; Lincoln, M.; Van Laar, B. Development and validation of a plasma-based melanoma biomarker suitable for clinical use. Br. J. Cancer 2018, 118, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumford, S.L.; Towler, B.P.; Pashler, A.L.; Gilleard, O.; Martin, Y.; Newbury, S.F. Circulating MicroRNA Biomarkers in Melanoma: Tools and Challenges in Personalised Medicine. Biomolecules 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Guo, W.; Li, S.; Dai, W.; Zhang, N.; Zhao, T.; Wang, H.; Ma, J.; Yi, X.; Ge, R.; et al. Se-rum miR-16: A Potential Biomarker for Predicting Melanoma Prognosis. J. Investig. Dermatol. 2016, 136, 985–993. [Google Scholar] [CrossRef]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBio Med. 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Katsuura, S.; Kuwano, Y.; Yamagishi, N.; Kurokawa, K.; Kajita, K.; Akaike, Y.; Nishida, K.; Masuda, K.; Tanahashi, T.; Rokutan, K. MicroRNAs miR-144/144* and miR-16 in peripheral blood are potential biomarkers for naturalistic stress in healthy Japanese medical students. Neurosci. Lett. 2012, 516, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
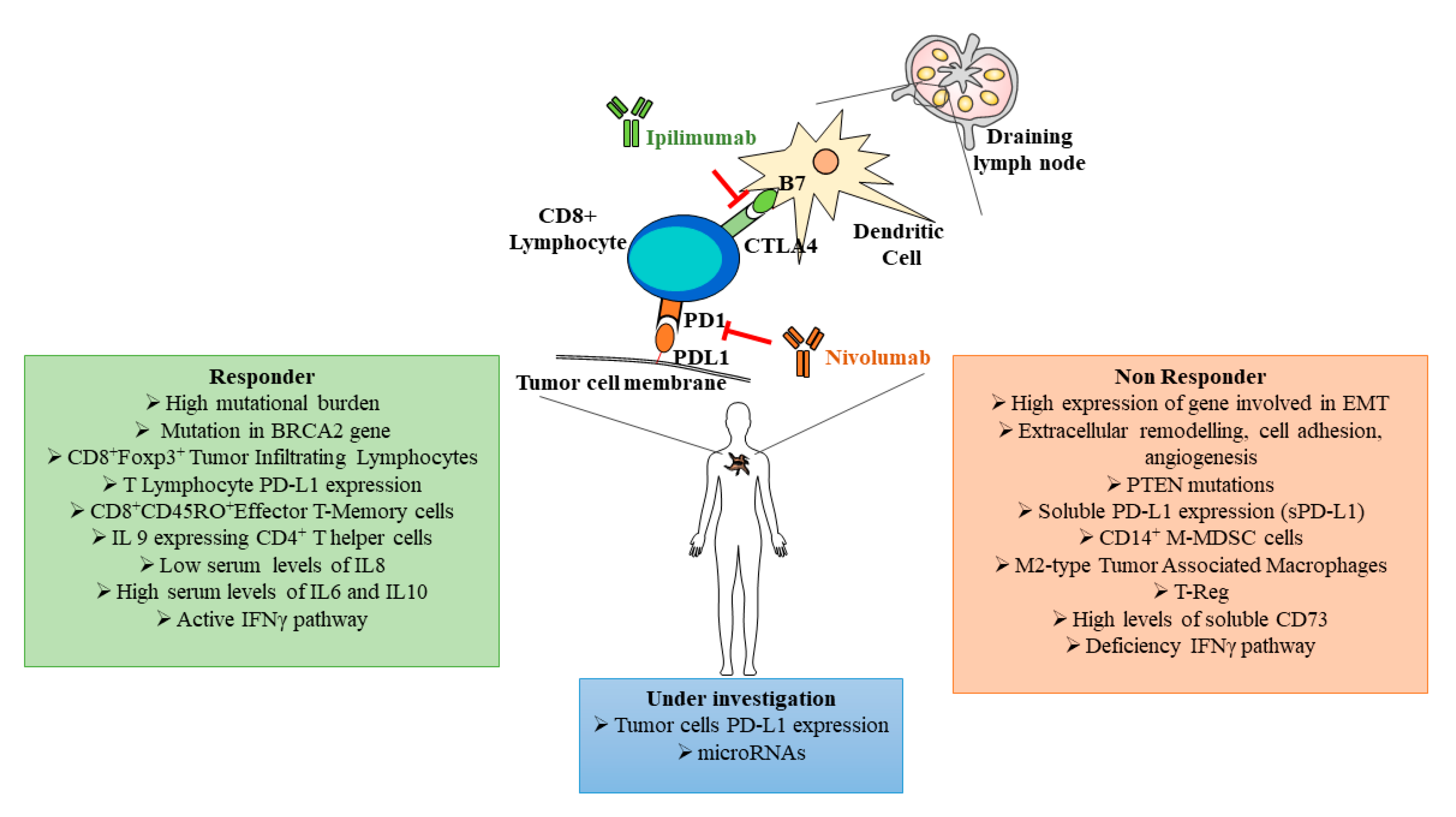{kind=link}
| Cellular Markers | Activity | References |
| CD8+Foxp3+ TILs | Antitumoral acitivity | [44] |
| CD8+ T Cytotoxic cells | Antigen primed cells against tumor cells, encountering dysfunction and exhaustion due to immunosuppression | [46] |
| CD14+ M-MDSCs | Cell population that inhibits T cell activation | [66] |
| M2-Type TAMs | Negatively modulates the antitumor T lymphocyte activity | [67] |
| T-reg | Cells that produce cytokines with immunosuppressive activities | [68] |
| CD8+CD45RO+ cells | T Memory cell subset | [69] |
| CD4+ Th-cells | Secreting cytokines with differential activities on other immune system cells | [70] |
| Circulating Molecules | Activity | References |
| IL-9 | Anti-tumoral actions in melanoma, increases granzyme B and perforin in CD8+ T cells | [70] |
| IL-8 | Key neutrophil chemotactic factor inducing chemotaxis and phagocytosis of target cells | [71] |
| IL-6 | Key pleiotropic cytokine with pro-tumorigenic and anti-tumoral role | [72] |
| IL-10 | Key immune-suppressive cytokine produced by T-reg | [72] |
| IFNγ | Cytokine playing an important role in inducing and modulating an array of immune responses | [72] |
| sCD73 | Participates in the extracellular production of adenosine that down-regulates inflammatory and immune responses | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puglisi, R.; Bellenghi, M.; Pontecorvi, G.; Pallante, G.; Carè, A.; Mattia, G. Biomarkers for Diagnosis, Prognosis and Response to Immunotherapy in Melanoma. Cancers 2021, 13, 2875. https://doi.org/10.3390/cancers13122875
Puglisi R, Bellenghi M, Pontecorvi G, Pallante G, Carè A, Mattia G. Biomarkers for Diagnosis, Prognosis and Response to Immunotherapy in Melanoma. Cancers. 2021; 13(12):2875. https://doi.org/10.3390/cancers13122875
Chicago/Turabian StylePuglisi, Rossella, Maria Bellenghi, Giada Pontecorvi, Giulia Pallante, Alessandra Carè, and Gianfranco Mattia. 2021. "Biomarkers for Diagnosis, Prognosis and Response to Immunotherapy in Melanoma" Cancers 13, no. 12: 2875. https://doi.org/10.3390/cancers13122875
APA StylePuglisi, R., Bellenghi, M., Pontecorvi, G., Pallante, G., Carè, A., & Mattia, G. (2021). Biomarkers for Diagnosis, Prognosis and Response to Immunotherapy in Melanoma. Cancers, 13(12), 2875. https://doi.org/10.3390/cancers13122875