
DNA Methylation Markers from Negative Surgical Margins Can Predict Recurrence of Oral Squamous Cell Carcinoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Sample Preparation for Methylome Profiling
2.3. DNA Methylome Profiling
2.4. Data Pre-Processing and Normalization
2.5. Differential DNAme Analysis
2.6. TCGA Replication
2.7. Assessment of Recurrence Markers
2.8. Survival Analysis
3. Results
3.1. Clinicopathological Characteristics
3.2. Identifying Recurrence-Associated DMPs
3.3. Identifying Recurrence-Associated DMRs
3.4. Identification of Potential Markers of OSCC Recurrence
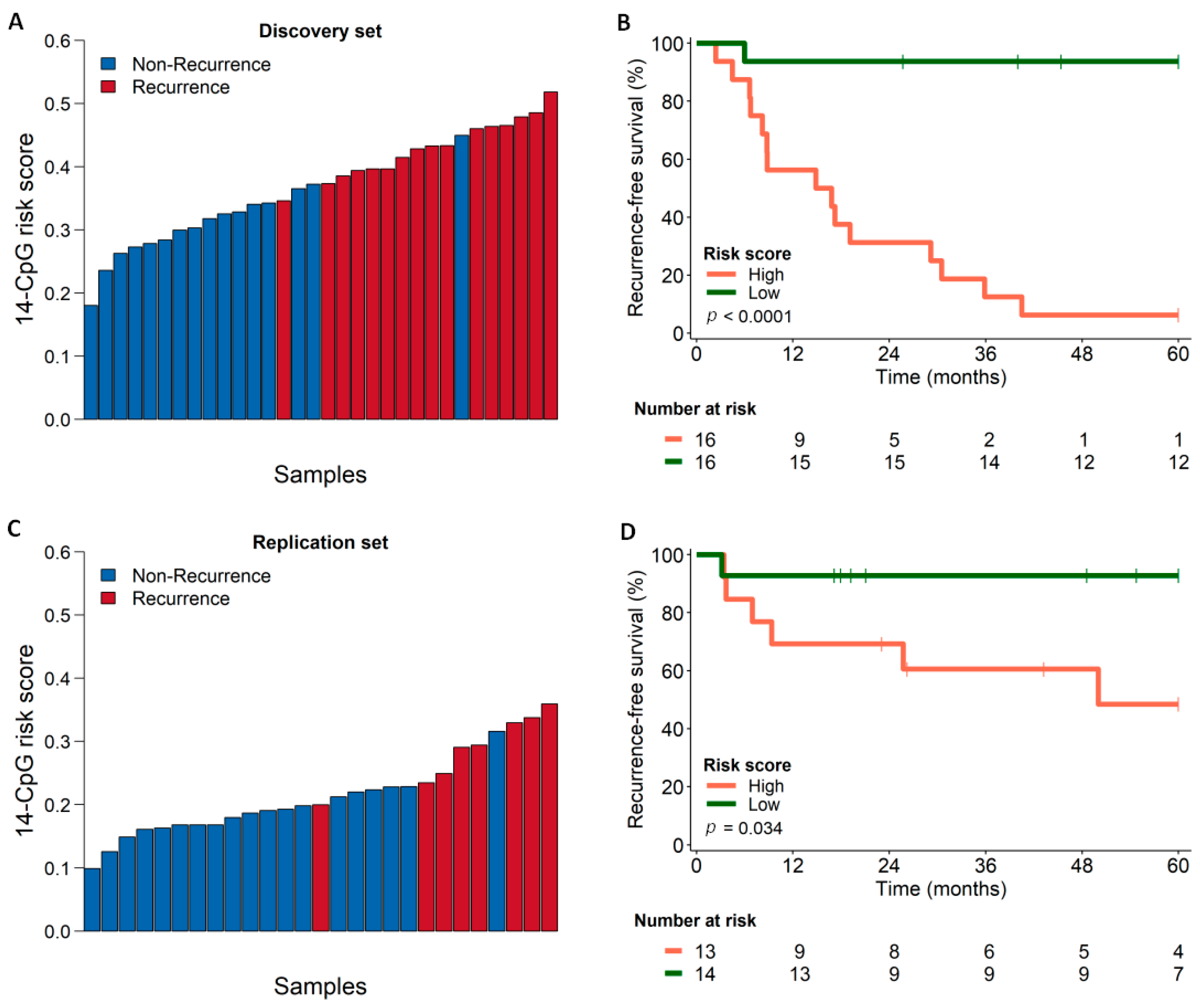
3.5. Survival Analysis Using Combined Score Generated from the Identified Marker Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Thompson, L.D.R. Head and neck cancers. In World Cancer Report 2014; Stewart, B.W., Wild, C.P., Eds.; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. Semin. Cancer Biol. 2020, 61, 71–83. [Google Scholar] [CrossRef]
- Rupel, K.; Ottaviani, G.; Gobbo, M.; Poropat, A.; Zoi, V.; Zacchigna, S.; Di Lenarda, R.; Biasotto, M. Campaign to Increase Awareness of Oral Cancer Risk Factors among Preadolescents. J. Cancer Educ. 2020, 35, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Sigston, E.A.; Longano, A.; Strzelecki, A.T.; Williams, B.R. Surgical margins in head and neck squamous cell carcinoma: Effect of heat artifact on immunohistochemistry as a future tool for assessment. Head Neck 2016, 38, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.G.; Spencer, S.; Adelstein, D.; Adkins, D.; Anzai, Y.; Brizel, D.M.; Bruce, J.Y.; Busse, P.M.; Caudell, J.J.; Cmelak, A.J.; et al. Head and Neck Cancers, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. JNCCN 2020, 18, 873–898. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Nishimoto, I.N.; Califano, J.A.; Kowalski, L.P. Trends in incidence and prognosis for head and neck cancer in the United States: A site-specific analysis of the SEER database. Int. J. Cancer 2005, 114, 806–816. [Google Scholar] [CrossRef]
- Surveillance, Epidemiology, and End Results (SEER) Program. Available online: https://seer.cancer.gov/statfacts/ (accessed on 14 July 2020).
- Reis, P.P.; Waldron, L.; Perez-Ordonez, B.; Pintilie, M.; Galloni, N.N.; Xuan, Y.; Cervigne, N.K.; Warner, G.C.; Makitie, A.A.; Simpson, C.; et al. A gene signature in histologically normal surgical margins is predictive of oral carcinoma recurrence. BMC Cancer 2011, 11, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Chang, J.H.; Wu, C.C.; Yuan, K.S.; Wu, A.T.H.; Wu, S.Y. Locoregionally recurrent head and neck squamous cell carcinoma: Incidence, survival, prognostic factors, and treatment outcomes. Oncotarget 2017, 8, 55600–55612. [Google Scholar] [CrossRef] [Green Version]
- Camisasca, D.R.; Silami, M.A.; Honorato, J.; Dias, F.L.; de Faria, P.A.; Lourenco Sde, Q. Oral squamous cell carcinoma: Clinicopathological features in patients with and without recurrence. ORL 2011, 73, 170–176. [Google Scholar] [CrossRef]
- Hayashi, M.; Wu, G.; Roh, J.L.; Chang, X.; Li, X.; Ahn, J.; Goldsmith, M.; Khan, Z.; Bishop, J.; Zhang, Z.; et al. Correlation of gene methylation in surgical margin imprints with locoregional recurrence in head and neck squamous cell carcinoma. Cancer 2015, 121, 1957–1965. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, A.C.; Kowalski, L.P.; Campos, A.H.; Soares, F.A.; Carvalho, A.L.; Vettore, A.L. Clinical significance of molecular alterations in histologically negative surgical margins of head and neck cancer patients. Oral Oncol. 2012, 48, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Chinn, S.B.; Myers, J.N. Oral Cavity Carcinoma: Current Management, Controversies, and Future Directions. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3269–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, A.R.; Greskovich, J.F.; Han, J.; Reddy, C.A.; Nwizu, T.I.; Khan, M.F.; Scharpf, J.; Adelstein, D.J.; Burkey, B.B.; Koyfman, S.A. Risk Factors Associated with Disease Recurrence in Patients with Stage III/IV Squamous Cell Carcinoma of the Oral Cavity Treated with Surgery and Postoperative Radiotherapy. Anticancer Res. 2016, 36, 785–792. [Google Scholar] [PubMed]
- Li, Y.F.; Hsiao, Y.H.; Lai, Y.H.; Chen, Y.C.; Chen, Y.J.; Chou, J.L.; Chan, M.W.; Lin, Y.H.; Tsou, Y.A.; Tsai, M.H.; et al. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics 2015, 10, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gissi, D.B.; Fabbri, V.P.; Gabusi, A.; Lenzi, J.; Morandi, L.; Melotti, S.; Asioli, S.; Tarsitano, A.; Balbi, T.; Marchetti, C.; et al. Pre-Operative Evaluation of DNA Methylation Profile in Oral Squamous Cell Carcinoma Can Predict Tumor Aggressive Potential. Int. J. Mol. Sci. 2020, 21, 6691. [Google Scholar] [CrossRef] [PubMed]
- Ha, P.K.; Califano, J.A. The molecular biology of mucosal field cancerization of the head and neck. Crit. Rev. Oral Biol. Med. Off. Publ. Am. Assoc. Oral Biol. 2003, 14, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Clark, D. Molecular margin of surgical resections--where do we go from here? Cancer 2015, 121, 1914–1916. [Google Scholar] [CrossRef]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]
- Wu, M.C.; Kuan, P.F. A Guide to Illumina BeadChip Data Analysis. Methods Mol. Biol. 2018, 1708, 303–330. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.L.; Dozmorov, M.G.; Wolen, A.R.; Jackson-Cook, C.; Starkweather, A.R.; Lyon, D.E.; York, T.P. Establishing an analytic pipeline for genome-wide DNA methylation. Clin. Epigenetics 2016, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef]
- KD, H. IlluminaHumanMethylationEPICanno.ilm10b4.hg19: Annotation for Illumina’s EPIC Methylation Arrays. R Package Version 0.6.0. Available online: https://bioconductor.org/packages/IlluminaHumanMethylationEPICanno.ilm10b4.hg19/ (accessed on 30 September 2020).
- KD, H. IlluminaHumanMethylation450kanno.ilmn12.hg19: Annotation for Illumina’s 450k Methylation Arrays. R Package Version 0.6.0. Available online: https://bioconductor.org/packages/IlluminaHumanMethylation450kanno.ilmn12.hg19/ (accessed on 30 September 2020).
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Perez-Enciso, M.; Tenenhaus, M. Prediction of clinical outcome with microarray data: A partial least squares discriminant analysis (PLS-DA) approach. Hum. Genet. 2003, 112, 581–592. [Google Scholar] [CrossRef]
- Talukdar, F.R.; Soares Lima, S.C.; Khoueiry, R.; Laskar, R.S.; Cuenin, C.; Sorroche, B.P.; Boisson, A.C.; Abedi-Ardekani, B.; Carreira, C.; Menya, D.; et al. Genome-Wide DNA Methylation Profiling of Esophageal Squamous Cell Carcinoma from Global High-Incidence Regions Identifies Crucial Genes and Potential Cancer Markers. Cancer Res. 2021, 81, 2612–2624. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alboukadel, K.; Marcin, K.; Przemyslaw, B.; Scheipl, F. Survminer: Drawing Survival Curves Using ‘ggplot2’. Available online: https://CRAN.R-project.org/package=survminer (accessed on 30 April 2020).
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef] [PubMed]
- Lever, J.; Zhao, E.Y.; Grewal, J.; Jones, M.R.; Jones, S.J.M. CancerMine: A literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat. Methods 2019, 16, 505–507. [Google Scholar] [CrossRef]
- Clark, D.J.; Mao, L. Understanding the Surgical Margin: A Molecular Assessment. Oral Maxillofac. Surg. Clin. N. Am. 2017, 29, 245–258. [Google Scholar] [CrossRef]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Bloemena, E.; Leemans, C.R.; Brakenhoff, R.H. Molecular analysis of surgical margins in head and neck cancer: More than a marginal issue. Oral Oncol. 2010, 46, 485–491. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk, L.; Owczarek, A.J. Methylation status of SFRP1, SFRP2, RASSF1A, RARbeta and DAPK1 genes in patients with oral squamous cell carcinoma. Arch. Oral Biol. 2019, 98, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Christensen, B.C.; Houseman, E.A.; Karagas, M.R.; Wrensch, M.R.; Yeh, R.F.; Nelson, H.H.; Wiemels, J.L.; Zheng, S.; Posner, M.R.; et al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis 2009, 30, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Demokan, S.; Dalay, N. Role of DNA methylation in head and neck cancer. Clin. Epigenetics 2011, 2, 123–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degli Esposti, D.; Sklias, A.; Lima, S.C.; Beghelli-de la Forest Divonne, S.; Cahais, V.; Fernandez-Jimenez, N.; Cros, M.P.; Ecsedi, S.; Cuenin, C.; Bouaoun, L.; et al. Unique DNA methylation signature in HPV-positive head and neck squamous cell carcinomas. Genome Med. 2017, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colacino, J.A.; Dolinoy, D.C.; Duffy, S.A.; Sartor, M.A.; Chepeha, D.B.; Bradford, C.R.; McHugh, J.B.; Patel, D.A.; Virani, S.; Walline, H.M.; et al. Comprehensive analysis of DNA methylation in head and neck squamous cell carcinoma indicates differences by survival and clinicopathologic characteristics. PLoS ONE 2013, 8, e54742. [Google Scholar] [CrossRef] [Green Version]
- Misawa, K.; Mochizuki, D.; Endo, S.; Mima, M.; Misawa, Y.; Imai, A.; Shinmura, K.; Kanazawa, T.; Carey, T.E.; Mineta, H. Site-specific methylation patterns of the GAL and GALR1/2 genes in head and neck cancer: Potential utility as biomarkers for prognosis. Mol. Carcinog. 2017, 56, 1107–1116. [Google Scholar] [CrossRef]
- Misawa, K.; Misawa, Y.; Kanazawa, T.; Mochizuki, D.; Imai, A.; Endo, S.; Carey, T.E.; Mineta, H. Epigenetic inactivation of galanin and GALR1/2 is associated with early recurrence in head and neck cancer. Clin. Exp. Metastasis 2016, 33, 187–195. [Google Scholar] [CrossRef]
- Oh, K.Y.; Hong, K.O.; Huh, Y.S.; Lee, J.I.; Hong, S.D. Decreased expression of SOX7 induces cell proliferation and invasion and correlates with poor prognosis in oral squamous cell carcinoma. J. Oral Pathol. Med. Off. Publ. Int. Assoc. Oral Pathol. Am. Acad. Oral Pathol. 2017, 46, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, G.; Shi, Q.; Zhang, R.; Zhao, Y.; Wei, Y.; Chen, F.; Christiani, D.C. Seven-CpG-based prognostic signature coupled with gene expression predicts survival of oral squamous cell carcinoma. Clin. Epigenetics 2017, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Mee, B.C.; Carroll, P.; Donatello, S.; Connolly, E.; Griffin, M.; Dunne, B.; Burke, L.; Flavin, R.; Rizkalla, H.; Ryan, C.; et al. Maintaining Breast Cancer Specimen Integrity and Individual or Simultaneous Extraction of Quality DNA, RNA, and Proteins from Allprotect-Stabilized and Nonstabilized Tissue Samples. Biopreserv. Biobank. 2011, 9, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.Y.; Hwang, K.S.; Yu, M.H.; Lee, H.S.; Lai, H.C.; Liu, J.Y. A research-based tumor tissue bank of gynecologic oncology: Characteristics of nucleic acids extracted from normal and tumor tissues from different sites. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2002, 12, 171–176. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Discovery Set n (%) | Replication Set n (%) |
|---|---|---|
| Mean age (range) | 62 (45–89) | 59.6 (26–80) |
| Gender | ||
| Male | 22 (68.8) | 22 (81.5) |
| Female | 10 (31.3) | 5 (18.5) |
| Tobacco Consumption | ||
| Never | 5 (15.6) | 6 (23.1) |
| Ever | 27 (84.4) | 20 (76.9) |
| Alcohol Consumption | ||
| Never | 9 (28.1) | 6 (23.1) |
| Ever | 23 (71.9) | 20 (76.9) |
| Anatomical Site | ||
| Floor of mouth | 11 (34.4) | 2 (7.4) |
| Tongue | 18 (56.3) | 13 (48.2) |
| Gingiva | 3 (9.4) | 1 (3.7) |
| Larynx | 0 (0.0) | 10 (37.0) |
| Base of tongue | 0 (0.0) | 1 (3.7) |
| Tumor Stage | ||
| I/II | 19 (59.4) | 2 (7.4) |
| III/IV | 13 (40.6) | 25 (92.6) |
| CpG-ID | Chr | Start | End | RefGene | Genomic Location | p-Value | Average β-Values | Δβ | |
|---|---|---|---|---|---|---|---|---|---|
| R | NR | ||||||||
| cg09830162 | 16 | 1,889,615 | 1,889,614 | FAHD1 | 3′UTR | <0.001 | 0.437 | 0.562 | −0.125 |
| cg04504441 | 5 | 132,948,218 | 132,948,217 | FSTL4 | 1st exon | <0.001 | 0.459 | 0.540 | −0.081 |
| cg05467458 | 19 | 33,361,033 | 33,361,032 | SLC7A9 | TSS1500 | 0.002 | 0.555 | 0.450 | 0.106 |
| cg07969676 | 8 | 10,590,642 | 10,590,641 | SOX7 | 5′UTR | 0.002 | 0.548 | 0.451 | 0.097 |
| cg03078363 | 12 | 54,408,665 | 54,408,664 | HOXC4 | TSS1500 | 0.002 | 0.541 | 0.460 | 0.081 |
| cg01580782 | 5 | 927,438 | 927,437 | non-genic | intergenic | 0.002 | 0.529 | 0.470 | 0.059 |
| cg03659519 | 18 | 74,961,967 | 74,961,966 | GALR1 | TSS200 | 0.005 | 0.570 | 0.430 | 0.141 |
| cg26252794 | 14 | 88,097,222 | 88,097,221 | non-genic | intergenic | 0.006 | 0.526 | 0.474 | 0.053 |
| cg01364862 | 3 | 159,364,475 | 159,364,474 | IQCJ-SCHIP1 | Body | 0.006 | 0.470 | 0.530 | −0.060 |
| cg14051544 | 3 | 170,303,287 | 170,303,286 | CLDN11 | 5′UTR | 0.007 | 0.556 | 0.447 | 0.109 |
| cg00363813 | 12 | 6,664,873 | 6,664,872 | IFFO1 | 1st exon | 0.016 | 0.554 | 0.446 | 0.108 |
| cg10094616 | 8 | 53,478,024 | 53,478,023 | ALKAL1 | TSS200 | 0.018 | 0.543 | 0.456 | 0.087 |
| cg14884793 | 13 | 109,148,554 | 109,148,553 | non-genic | Intergenic | 0.019 | 0.534 | 0.466 | 0.068 |
| cg00082235 | 12 | 6,664,537 | 6,664,536 | IFFO1 | 1st exon | 0.032 | 0.538 | 0.462 | 0.076 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorroche, B.P.; Talukdar, F.R.; Lima, S.C.S.; Melendez, M.E.; de Carvalho, A.C.; de Almeida, G.C.; De Marchi, P.; Lopes, M.; Ribeiro Pinto, L.F.; Carvalho, A.L.; et al. DNA Methylation Markers from Negative Surgical Margins Can Predict Recurrence of Oral Squamous Cell Carcinoma. Cancers 2021, 13, 2915. https://doi.org/10.3390/cancers13122915
Sorroche BP, Talukdar FR, Lima SCS, Melendez ME, de Carvalho AC, de Almeida GC, De Marchi P, Lopes M, Ribeiro Pinto LF, Carvalho AL, et al. DNA Methylation Markers from Negative Surgical Margins Can Predict Recurrence of Oral Squamous Cell Carcinoma. Cancers. 2021; 13(12):2915. https://doi.org/10.3390/cancers13122915
Chicago/Turabian StyleSorroche, Bruna Pereira, Fazlur Rahman Talukdar, Sheila Coelho Soares Lima, Matias Eliseo Melendez, Ana Carolina de Carvalho, Gisele Caravina de Almeida, Pedro De Marchi, Monique Lopes, Luis Felipe Ribeiro Pinto, André Lopes Carvalho, and et al. 2021. "DNA Methylation Markers from Negative Surgical Margins Can Predict Recurrence of Oral Squamous Cell Carcinoma" Cancers 13, no. 12: 2915. https://doi.org/10.3390/cancers13122915