
Dual Role of Integrin Alpha-6 in Glioblastoma: Supporting Stemness in Proneural Stem-Like Cells While Inducing Radioresistance in Mesenchymal Stem-Like Cells

 , , ,
, , ,  , and
, and 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Cell Lines and Differentiation Assay
2.2. RNA Isolation and Real-Time q-PCR Analyses
2.3. Flow Cytometry and Integrin a6 Cell Sorting
2.4. Western Blot
2.5. Gliomasphere Formation Assay
2.6. Lentiviral Particle Generation and GSCs Transduction
2.7. Radiation Schedule
2.8. Gamma-H2AX Foci Formation Assay
2.9. Clonogenic Assay and Linear-Quadratic Model Interpretation
2.10. In-Vitro Extreme Limiting Dilution Assay (ELDA)
2.11. Bulk RNA-Seq and Bioinformatic Analysis
2.12. Elaboration of Publicly Available Data from GBM Patients
2.13. Statistical Analysis
3. Results
3.1. Integrin a6 Is Expressed in Proneural and Mesenchymal GBM Subtypes
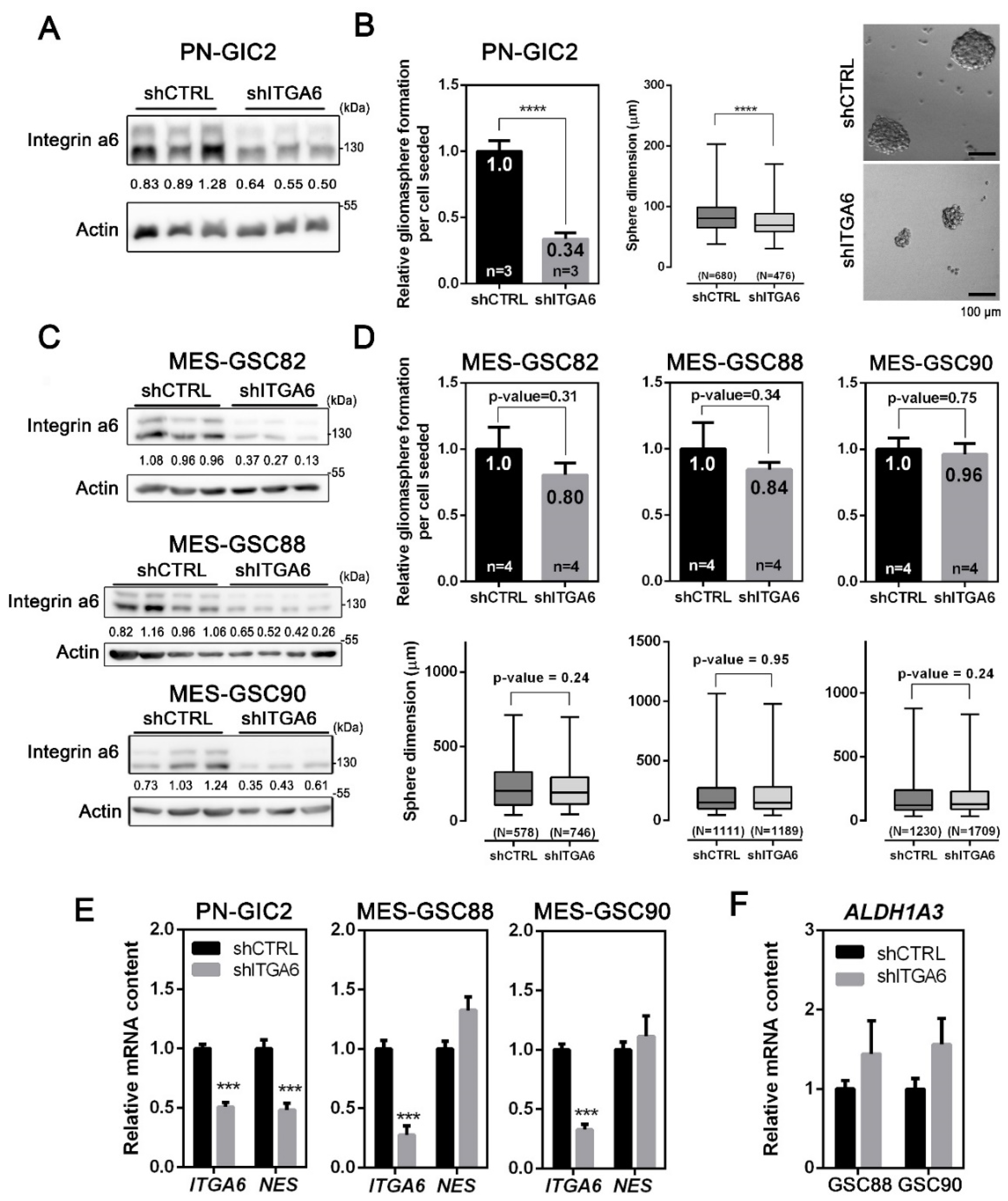
3.2. Integrin a6 Supports Stemness in PN-GSCs, but Not in MES-GSCs
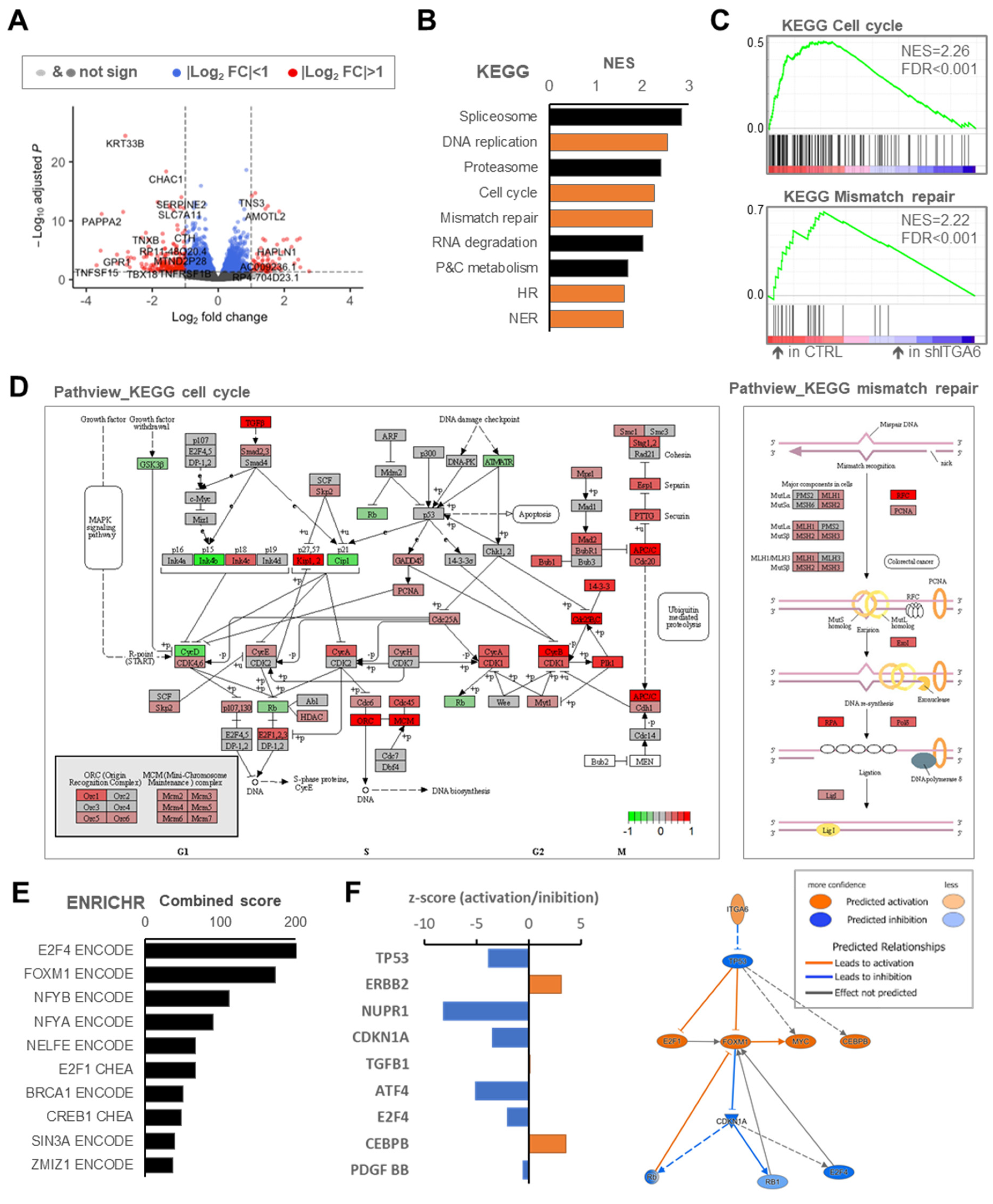
3.3. Downregulation of Integrin a6 Affects DNA Damage Repair and Cell Cycle in MES-GSCs
3.4. Integrin a6 Is Critical for MES-GSCs Sensitivity to Ionizing Radiation
4. Discussion
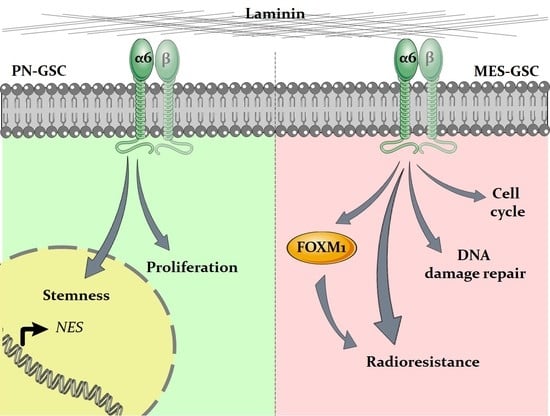
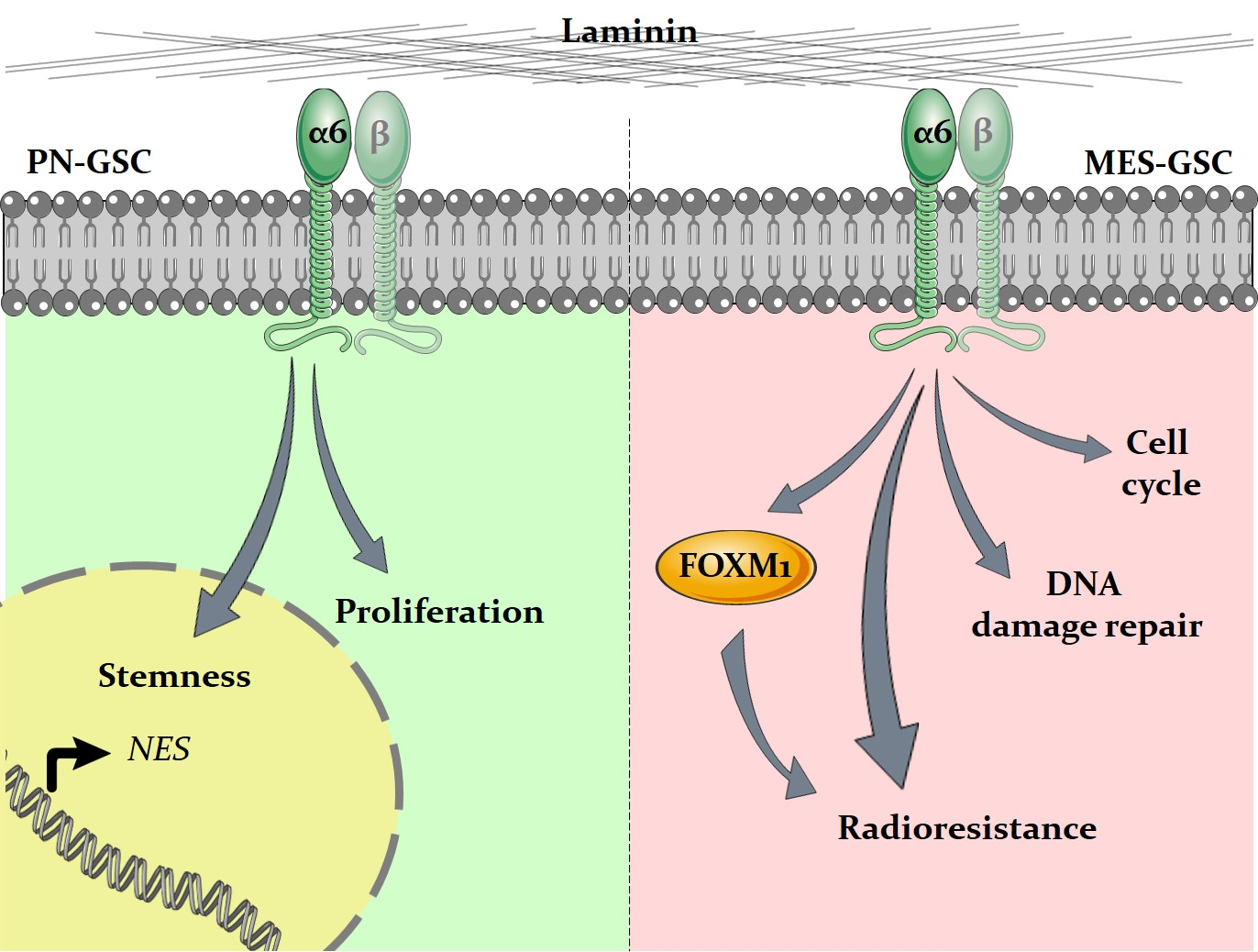
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of Glioblastoma: State of the Art and Future Directions. CA. Cancer J. Clin. 2020, 70, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Laperriere, N.; Zuraw, L.; Cairncross, G.; Cancer, T.; Ontario, C. Radiotherapy for newly diagnosed malignant glioma in adults: A systematic review. Radiother. Oncol. 2002, 64, 259–273. [Google Scholar] [CrossRef]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016; ISBN 9789283244929. [Google Scholar]
- Lathia, J.; Mack, S.; Mulkearns-Hubert, E.E.; Valentim, C.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Seano, G. Targeting the perivascular niche in brain tumors. Curr. Opin. Oncol. 2018, 30, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends in Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, R.; Ahmed, S.U.; Strathdee, K.; Gomez-Roman, N.; Amoah-Buahin, E.; Watts, C.; Chalmers, A.J. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol. Oncol. 2015, 9, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Caragher, S.P.; Sachdev, S.; Ahmed, A.U. Radiotherapy and Glioma Stem Cells: Searching for Chinks in Cellular Armor. Curr. Stem Cell Reports 2017, 3, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends in Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2019, 11, 1–17. [Google Scholar] [CrossRef]
- Vescovi, A.L.; Galli, R.; Reynolds, B. A Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, D.; Pouwels, J.; Franceschi, N.D.; Ivaska, J. Integrin inactivators: Balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 2013, 14, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.J.; Tanentzapf, G. Integrin-mediated adhesion and stem-cell-niche interactions. Cell Tissue Res. 2010, 339, 121–130. [Google Scholar] [CrossRef]
- Bigoni-Ordóñez, G.D.; Czarnowski, D.; Parsons, T.; Madlambayan, G.J.; Villa-Diaz, L.G. Integrin α6 (CD49f), The Microenvironment and Cancer Stem Cells. Curr. Stem Cell Res. Ther. 2019, 14, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Malric, L.; Monferran, S.; Gilhodes, J.; Boyrie, S.; Dahan, P.; Skuli, N.; Sesen, J.; Filleron, T.; Kowalski-Chauvel, A.; Moyal, E.C.J.; et al. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: An update. Oncotarget 2017, 8, 86947–86968. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-R.; Yang, S.-R.; Jung, J.-W.; Kim, H.; Ko, K.; Han, D.W.; Park, S.-B.; Choi, S.W.; Kang, S.-K.; Schöler, H.; et al. CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells 2012, 30, 876–887. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Kowalski-Chauvel, A.; Modesto, A.; Gouaze-andersson, V.; Baricault, L.; Gilhodes, J.; Delmas, C.; Lemarie, A.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. Alpha-6 integrin promotes radioresistance of glioblastoma by modulating DNA damage response and the transcription factor Zeb1. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.a.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.-H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic Plasticity of Invasive Edge Glioma Stem-like Cells in Response to Ionizing Radiation. Cell Rep. 2019, 26, 1893–1904.e8. [Google Scholar] [CrossRef]
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Goel, H.L.; Gritsko, T.; Pursell, B.; Chang, C.; Shultz, L.D.; Greiner, D.L.; Norum, J.H.; Toftgard, R.; Shaw, L.M.; Mercurio, A.M. Regulated Splicing of the α6 Integrin Cytoplasmic Domain Determines the Fate of Breast Cancer Stem Cells. Cell Rep. 2014, 7, 747–761. [Google Scholar] [CrossRef]
- Stanzani, E.; Martínez-Soler, F.; Martín Mateos, T.; Vidal, N.; Villanueva, A.; Pujana, M.A.; Serra-Musach, J.; de la Iglesia, N.; Giménez-Bonafé, P.; Tortosa, A. Radioresistance of mesenchymal glioblastoma initiating cells correlates with patient outcome and is associated with activation of inflammatory program. Oncotarget 2017, 8, 73640–73653. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Gritti, A.; Parati, E.; Cova, L.; Frolichsthal, P.; Galli, R.; Wanke, E.; Faravelli, L.; Morassutti, D.; Roisen, F.; Nickel, D.; et al. Multipotential Stem Cells from the Adult Mouse and Self-Renew in Response to Basic Fibroblast. J. Neurosci. 1996, 16, 1091–1100. [Google Scholar] [CrossRef]
- Franken, N.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J. The linear-quadratic model is an appropriate methodology for determining isoeffective doses at large doses per fraction. Semin. Radiat. Oncol. 2008, 18, 234–239. [Google Scholar] [CrossRef]
- Franken, N.; van Bree, C.; ten Cate, R.; van Oven, C.H.; Haveman, J. Importance of TP53 and RB in the repair of potentially lethal damage and induction of color junctions after exposure to ionizing radiation. Radiat. Res. 2002, 158, 707–714. [Google Scholar] [CrossRef]
- De Llobet, L.I.; Baro, M.; Figueras, A.; Modolell, I.; Da Silva, M.V.; Muñoz, P.; Navarro, A.; Mesia, R.; Balart, J. Development and characterization of an isogenic cell line with a radioresistant phenotype. Clin. Transl. Oncol. 2013, 15, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- TCGA network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Madhavan, S.; Zenklusen, J.C.; Kotliarov, Y.; Sahni, H.; Fine, H.A.; Buetow, K. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 2009, 7, 157–167. [Google Scholar] [CrossRef]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.C.; Achrol, A.S.; Januszyk, M.; Kahn, S.A.; Liu, T.T.; Liu, Y.; Sahoo, D.; Rodrigues, M.; Maan, Z.N.; Wong, V.W.; et al. Multiple Subsets of Brain Tumor Initiating Cells Coexist in Glioblastoma. Stem Cells 2016, 34, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Gagliardi, F.; Gallotti, A.L.; Mazzoleni, S.; Cominelli, M.; Fagnocchi, L.; Pala, M.; Piras, I.S.; Zordan, P.; Moretta, N.; et al. The proneural gene ASCL1 governs the transcriptional subgroup affiliation in glioblastoma stem cells by directly repressing the mesenchymal gene NDRG1. Cell Death Differ. 2019, 26, 1813–1831. [Google Scholar] [CrossRef]
- Mori, H.; Ninomiya, K.; Kino-oka, M.; Shofuda, T.; Islam, M.; Yamasaki, M.; Okano, H.; Taya, M.; Kanemura, Y. Effect of neurosphere size on the growth rate of Human Neural Stem/Progenitor Cells. J. Neurosci. Res. 2006, 84, 1682–1691. [Google Scholar] [CrossRef]
- Román-Trufero, M.; Méndez-Gómez, H.R.; Pérez, C.; Hijikata, A.; Fujimura, Y.I.; Endo, T.; Koseki, H.; Vicario-Abejón, C.; Vidal, M. Maintenance of undifferentiated state and self-renewal of embryonic neural stem cells by polycomb protein Ring1B. Stem Cells 2009, 27, 1559–1570. [Google Scholar] [CrossRef]
- Singec, I.; Knoth, R.; Meyer, R.P.; Maciaczyk, J.; Volk, B.; Nikkhah, G.; Frotscher, M.; Snyder, E.Y. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat. Methods 2006, 3, 801–806. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, G.; Sun, Y. A reliable parameter to standardize the scoring of stem cell spheres. PLoS ONE 2015, 10, 1–13. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef]
- Kowalski-Chauvel, A.; Gouaze-Andersson, V.; Baricault, L.; Martin, E.; Delmas, C.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. Alpha6-integrin regulates FGFR1 expression through the ZEB1/YAP1 transcription complex in glioblastoma stem cells resulting in enhanced proliferation and stemness. Cancers 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; O’Sullivan, H. FOXM1 in sarcoma: Role in cell cycle, pluripotency genes and stem cell pathways. Oncotarget 2016, 7, 42792–42804. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Gouazé-Andersson, V.; Ghérardi, M.J.; Lemarié, A.; Gilhodes, J.; Lubrano, V.; Arnauduc, F.; Moyal, E.C.J.; Toulas, C. FGFR1/FOXM1 pathway: A key regulator of glioblastoma stem cells radioresistance and a prognosis biomarker. Oncotarget 2018, 9, 31637–31649. [Google Scholar] [CrossRef]
- Maachani, U.B.; Shankavaram, U.; Kramp, T.; Tofilon, P.J.; Camphausen, K.; Tandle, A.T. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget 2016, 7, 77365–77377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wu, X.; Yang, L.; Xiao, F.; Zhang, H.; Zhou, A.; Huang, Z.; Huang, S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin. Cancer Res. 2012, 18, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, K.H.; Kim, D.G.; Cho, H.J.; Kim, Y.; Rheey, J.; Shin, K.; Seo, Y.J.; Choi, Y.S.; Lee, J.I.; et al. FoxM1 promotes stemness and radio- resistance of glioblastoma by regulating the master stem cell regulator Sox2. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef]
- Zona, S.; Bella, L.; Burton, M.J.; Nestal de Moraes, G.; Lam, E.W.F. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Allmann, S.; Mayer, L.; Olma, J.; Kaina, B.; Hofmann, T.G.; Tomicic, M.T.; Christmann, M. Benzo[a]pyrene represses DNA repair through altered E2F1/E2F4 function marking an early event in DNA damage-induced cellular senescence. Nucleic Acids Res. 2021, 48, 12085–12101. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.G.; Boulton, S.J. Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J. Practical time-dose evaluations, or how to stop worrying and learn to love linear quadratics. In Technical Basis of Radiation Therapy Medical Radiology (Radiation Oncology); Levitt, S.H., Purdy, J.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 3–50. ISBN 978-3-540-21338-3, 978-3-540-35665-3. [Google Scholar]
- Astrahan, M. Some implications of linear-quadratic-linear radiation dose-response with regard to hypofractionation. Med. Phys. 2008, 35, 4161–4172. [Google Scholar] [CrossRef] [PubMed]
- Emami, B.; Woloschak, G.; Small, W.J. Beyond the linear quadratic model: Intraoperative radiotherapy and normal tissue tolerance. Transl. Cancer Res. 2015, 4, 140–147. [Google Scholar] [CrossRef]
- Chapman, J.D. Single-hit mechanism of tumour cell killing by radiation. Int. J. Radiat. Biol. 2003, 79, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Primo, L.; Seano, G.; Roca, C.; Maione, F.; Gagliardi, P.A.; Sessa, R.; Martinelli, M.; Giraudo, E.; Di Blasio, L.; Bussolino, F. Increased expression of α6 integrin in endothelial cells unveils a proangiogenic role for basement membrane. Cancer Res. 2010, 70, 5759–5769. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Chiaverina, G.; Gagliardi, P.A.; Di Blasio, L.; Puliafito, A.; Bouvard, C.; Sessa, R.; Tarone, G.; Sorokin, L.; Helley, D.; et al. Endothelial podosome rosettes regulate vascular branching in tumour angiogenesis. Nat. Cell Biol. 2014, 16, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Mikkelsen, T.; Rosenfeld, S.S.; Hochberg, F.; Akella, N.S.; Fisher, J.D.; Cloud, G.A.; Zhang, Y.; Carson, K.; Wittemer, S.M.; et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J. Clin. Oncol. 2007, 25, 1651–1657. [Google Scholar] [CrossRef]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro. Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Löbrich, M.; Jeggo, P. A The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 2007, 7, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhou, R.; Zhao, Y.; Wu, G. Integrin alpha6/Akt/Erk signaling is essential for human breast cancer resistance to radiotherapy. Sci. Rep. 2016, 6, 33376. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Carla da Hora, C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, E.A.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro. Oncol. 2017, 19, now253. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2016 WHO classification of tumours of the central nervous system: The major points of revision. Neurol. Med. Chir. (Tokyo) 2017, 57, 301–311. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Giordana, M.T.; Germano, I.; Giaccone, G.; Mauro, A.; Migheli, A.; Schiffer, D. The distribution of laminin in human brain tumors: An immunohistochemical study. Acta Neuropathol. 1985, 67, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y. Laminin: Loss-of-function studies. Cell. Mol. Life Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, Y.; Mii, S.; Enomoto, A.; Momota, H.; Han, Y.P.; Kato, T.; Ushida, K.; Kato, A.; Asai, N.; Murakumo, Y.; et al. Significance of perivascular tumour cells defined by CD109 expression in progression of glioma. J. Pathol. 2017, 243, 468–480. [Google Scholar] [CrossRef]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; D’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma stem cells microenvironment: The paracrine roles of the niche in drug and radioresistance. Stem Cells Int. 2016, 2016, e6809105. [Google Scholar] [CrossRef]
- Silver, D.J.; Lathia, J.D. Revealing the glioma cancer stem cell interactome, one niche at a time. J. Pathol. 2018, 244, 260–264. [Google Scholar] [CrossRef]
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.S.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Perspective Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel co-option in glioblastoma: Emerging insights and opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanzani, E.; Pedrosa, L.; Bourmeau, G.; Anezo, O.; Noguera-Castells, A.; Esteve-Codina, A.; Passoni, L.; Matteoli, M.; de la Iglesia, N.; Seano, G.; et al. Dual Role of Integrin Alpha-6 in Glioblastoma: Supporting Stemness in Proneural Stem-Like Cells While Inducing Radioresistance in Mesenchymal Stem-Like Cells. Cancers 2021, 13, 3055. https://doi.org/10.3390/cancers13123055
Stanzani E, Pedrosa L, Bourmeau G, Anezo O, Noguera-Castells A, Esteve-Codina A, Passoni L, Matteoli M, de la Iglesia N, Seano G, et al. Dual Role of Integrin Alpha-6 in Glioblastoma: Supporting Stemness in Proneural Stem-Like Cells While Inducing Radioresistance in Mesenchymal Stem-Like Cells. Cancers. 2021; 13(12):3055. https://doi.org/10.3390/cancers13123055
Chicago/Turabian StyleStanzani, Elisabetta, Leire Pedrosa, Guillaume Bourmeau, Oceane Anezo, Aleix Noguera-Castells, Anna Esteve-Codina, Lorena Passoni, Michela Matteoli, Núria de la Iglesia, Giorgio Seano, and et al. 2021. "Dual Role of Integrin Alpha-6 in Glioblastoma: Supporting Stemness in Proneural Stem-Like Cells While Inducing Radioresistance in Mesenchymal Stem-Like Cells" Cancers 13, no. 12: 3055. https://doi.org/10.3390/cancers13123055
APA StyleStanzani, E., Pedrosa, L., Bourmeau, G., Anezo, O., Noguera-Castells, A., Esteve-Codina, A., Passoni, L., Matteoli, M., de la Iglesia, N., Seano, G., Martínez-Soler, F., & Tortosa, A. (2021). Dual Role of Integrin Alpha-6 in Glioblastoma: Supporting Stemness in Proneural Stem-Like Cells While Inducing Radioresistance in Mesenchymal Stem-Like Cells. Cancers, 13(12), 3055. https://doi.org/10.3390/cancers13123055