Targeting the RhoGEF βPIX/COOL-1 in Glioblastoma: Proof of Concept Studies

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. βPix/COOL-1 Plays a Key Role in GBM Cell Invasion
2.2. βPix/COOL-1 Mediates GBM Cell Invasion
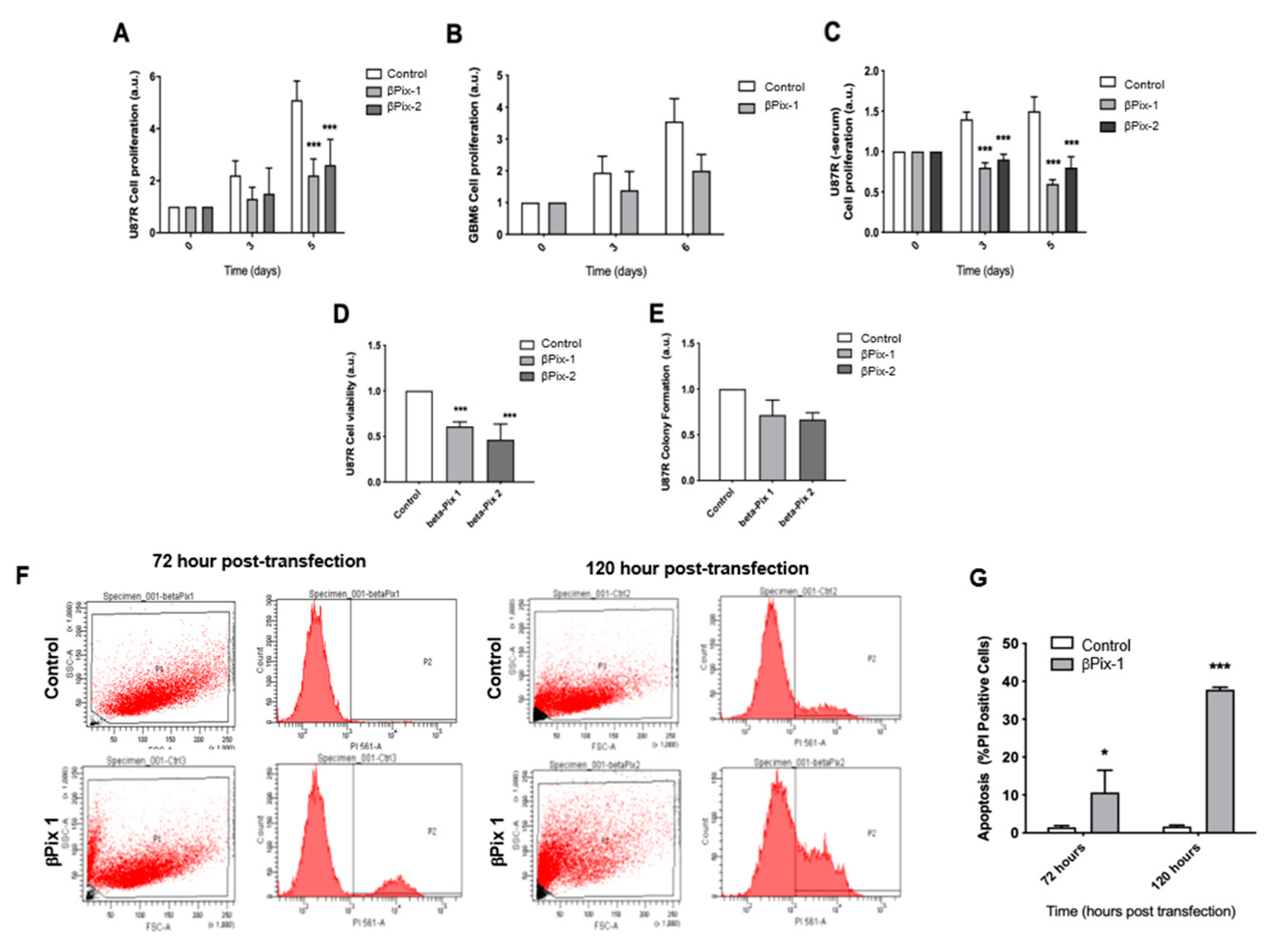
2.3. βPix/COOL-1 Knockdown Decreases GBM Proliferation and Viability and Enhances Apoptosis
2.4. βPix/COOL-1 Mediates Endothelial Cell Migration but Not Endothelial Tube Formation or Proliferation
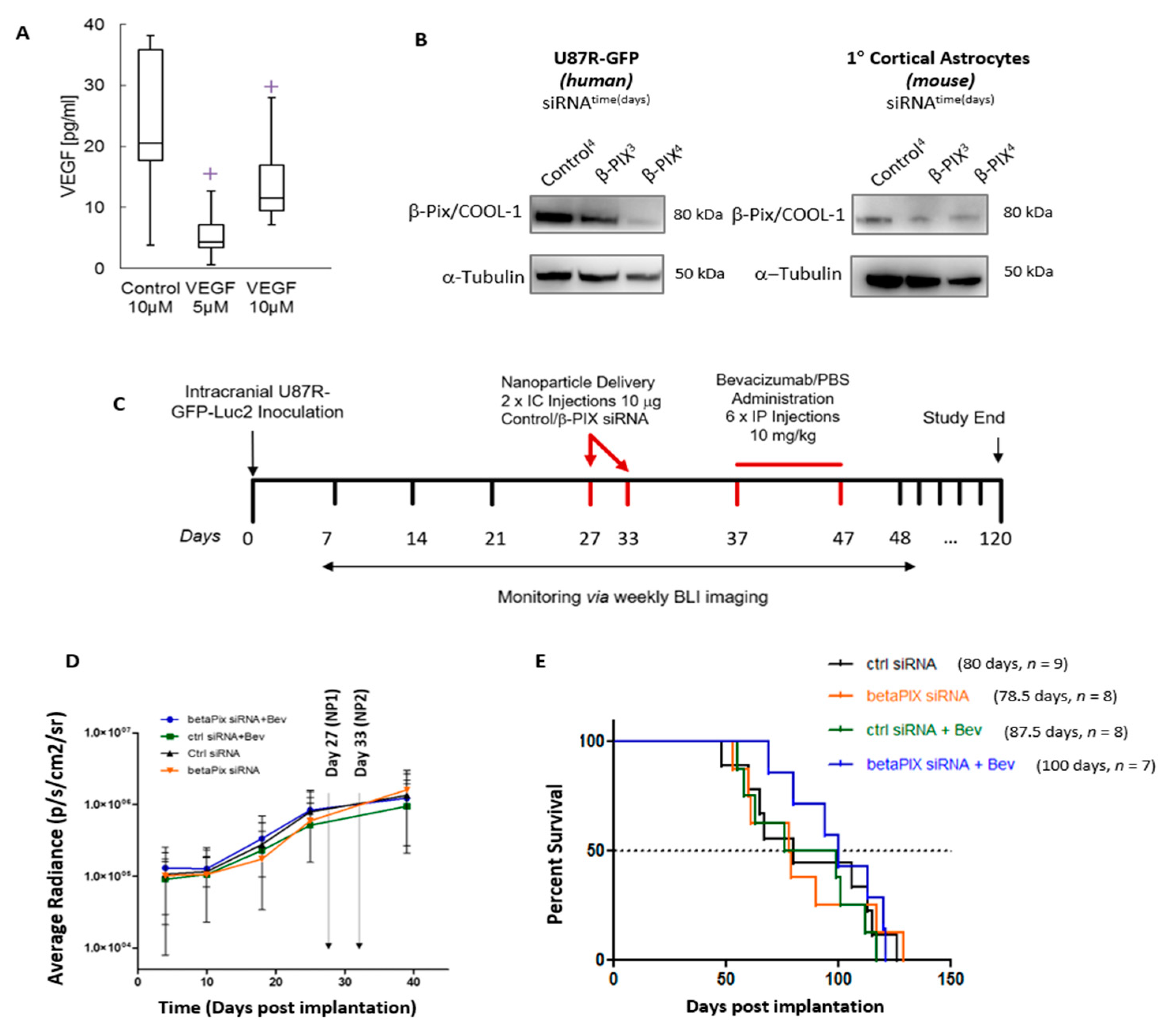
2.5. In Vivo Assessment of βPix/COOL-1 siRNA Loaded RGD-NP in Combination with Bev Indicates a Trend Towards Improved Survival
3. Discussion
4. Materials and Methods
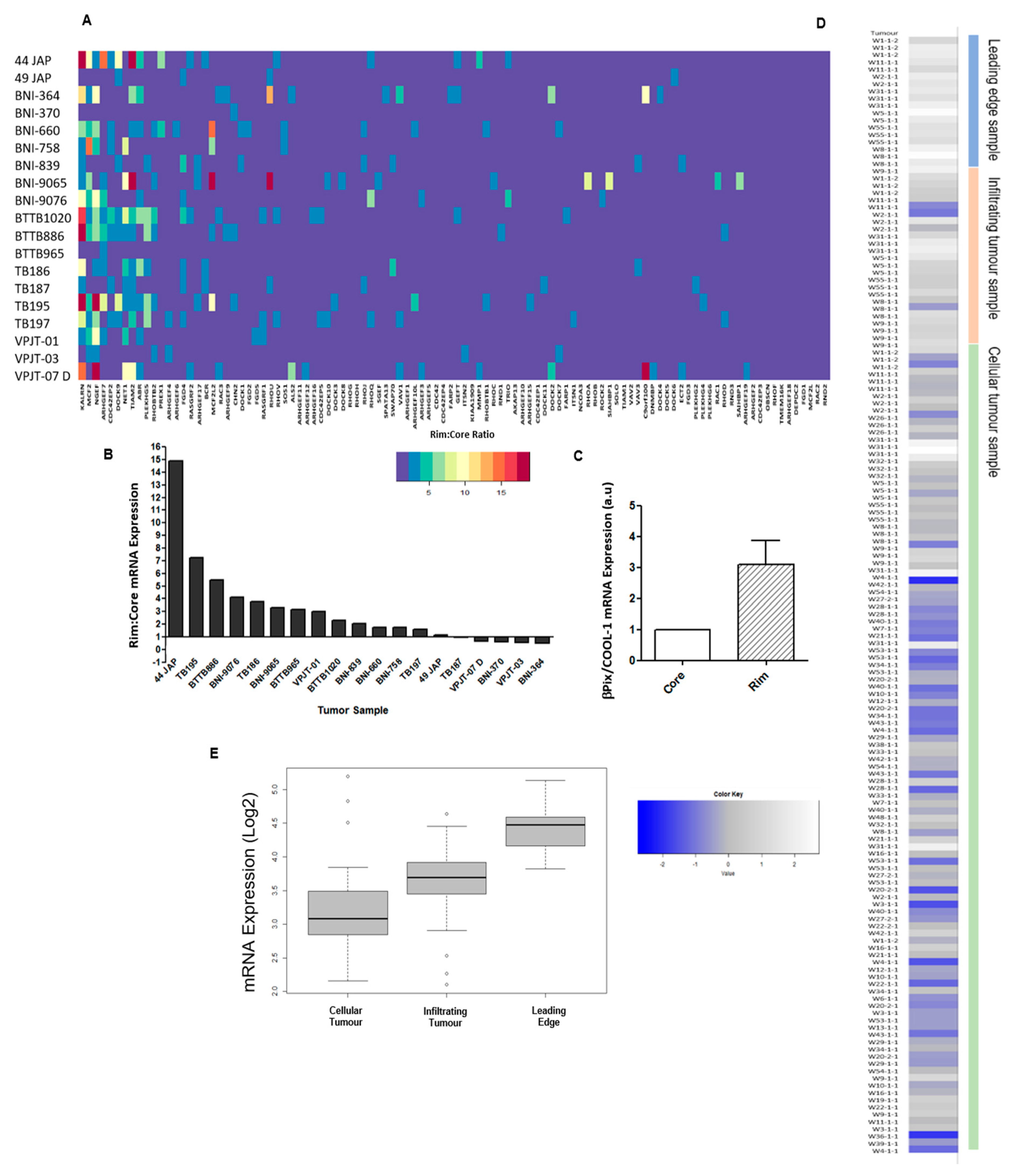
4.1. Identification of RhoGEFs Differentially Expressed in the Leading Edge of Patient Tumours
4.2. Cell Culture
4.3. siRNA Transfections
4.4. Western Blot Analysis of βPix/COOL-1 Expression
4.5. Sulpho-Rhodamine B (SRB) Proliferation Assay
4.6. Cell Cycle Analysis of GBM Cells
4.7. Matrigel Invasion Assay
4.8. Migration Assay
4.9. Clonogenic Assay
4.10. Tube Formation (Angiogenesis) Assay
4.11. Enzyme-Linked Immunosorbent Assay
4.12. GBM Orthoxenograft Studies
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.A.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.; van den Bent, M.J.; Weller, M.; Fisher, B.M.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef]
- Chi, A.S.; Sorensen, A.G.; Jain, R.K.; Batchelor, T.T. Angiogenesis as a Therapeutic Target in Malignant Gliomas. Oncologist 2009, 14, 621–636. [Google Scholar] [CrossRef] [Green Version]
- Liao, K.-L.; Huang, S.; Wu, Y.-P. The prognosis for patients with newly diagnosed glioblastoma receiving bevacizumab combination therapy: A meta-analysis. Onco Targets Ther. 2018, 11, 3513–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutsch, M.B.; Panageas, K.S.; Lassman, A.B.; DeAngelis, L.M. Steroid management in newly diagnosed glioblastoma. J. Neurooncol. 2013, 113, 111–116. [Google Scholar] [CrossRef]
- Kunkel, P.; Ulbricht, U.; Bohlen, P.; Brockmann, M.A.; Fillbrandt, R.; Stavrou, D.; Westphal, M.; Lamszus, K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001, 61, 6624–6628. [Google Scholar]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Ishida, J.; Kurozumi, K.; Ichikawa, T.; Otani, Y.; Oka, T.; Tomita, Y.; Hattori, Y.; Uneda, A.; Matsumoto, Y.; et al. δ-Catenin Promotes Bevacizumab-Induced Glioma Invasion. Mol. Cancer 2019, 18, 812–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucio-Eterovic, A.K.; Piao, Y.; de Groot, J.F. Mediators of Glioblastoma Resistance and Invasion during Antivascular Endothelial Growth Factor Therapy. Clin. Cancer Res. 2009, 15, 4589–4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahangiri, A.; De Lay, M.; Miller, L.M.; Carbonell, W.S.; Hu, Y.-L.; Lu, K.; Tom, M.W.; Paquette, J.; Tokuyasu, T.A.; Tsao, S.; et al. Gene Expression Profile Identifies Tyrosine Kinase c-Met as a Targetable Mediator of Antiangiogenic Therapy Resistance. Clin. Cancer Res. 2013, 19, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fack, F.; Espedal, H.; Keunen, O.; Golebiewska, A.; Obad, N.; Harter, P.N.; Mittelbronn, M.; Bähr, O.; Weyerbrock, A.; Stuhr, L.; et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 2015, 129, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, R.M.; Torcuator, R.; Jain, R.; Anderson, J.; Doyle, T.; Ellika, S.; Schultz, L.; Mikkelsen, T. Efficacy, safety and patterns of response and recurrence in patients with recurrent high-grade gliomas treated with bevacizumab plus irinotecan. J. Neurooncol. 2009, 91, 329–336. [Google Scholar] [CrossRef]
- De Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Fortin Ensign, S.P.; Mathews, I.T.; Symons, M.H.; Berens, M.E.; Tran, N.L. Implications of Rho GTPase Signaling in Glioma Cell Invasion and Tumor Progression. Front. Oncol. 2013, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Meel, R.; Symons, M.H.; Kudernatsch, R.; Kok, R.J.; Schiffelers, R.M.; Storm, G.; Gallagher, W.M.; Byrne, A.T. The VEGF/Rho GTPase signalling pathway: A promising target for anti-angiogenic/anti-invasion therapy. Drug Discov. Today 2011, 16, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.W.; Didier, S.; Chan, A.; Paulino, V.; Van Aelst, L.; Ruggieri, R.; Tran, N.L.; Byrne, A.T.; Symons, M. Guanine nucleotide exchange factor Dock7 mediates HGF-induced glioblastoma cell invasion via Rac activation. Br. J. Cancer 2014, 110, 1307–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Dhruv, H.; Kwiatkowska-Piwowarczyk, A.; Ruggieri, R.; Kloss, J.; Symons, M.; Pirrotte, P.; Eschbacher, J.M.; Tran, N.L.; Loftus, J.C. PDZ-RhoGEF Is a Signaling Effector for TROY-Induced Glioblastoma Cell Invasion and Survival. Neoplasia 2018, 20, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Dong, Z.; Yang, Y.; Fortin Ensign, S.P.; Sabit, H.; Nakada, M.; Ruggieri, R.; Kloss, J.M.; Symons, M.; Tran, N.L.; et al. Leukemia-Associated Rho Guanine Nucleotide Exchange Factor and Ras Homolog Family Member C Play a Role in Glioblastoma Cell Invasion and Resistance. Am. J. Pathol. 2020, 190, 2165–2176. [Google Scholar] [CrossRef]
- White, K.; Connor, K.; Clerkin, J.; Murphy, B.M.; Salvucci, M.; O’Farrell, A.C.; Rehm, M.; O’Brien, D.; Prehn, J.H.M.; Niclou, S.P.; et al. New hints towards a precision medicine strategy for IDH wild-type glioblastoma. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Voloshin, T.; Schneiderman, R.S.; Volodin, A.; Shamir, R.R.; Kaynan, N.; Zeevi, E.; Koren, L.; Klein-Goldberg, A.; Paz, R.; Giladi, M.; et al. Tumor Treating Fields (TTFields) Hinder Cancer Cell Motility through Regulation of Microtubule and Actin Dynamics. Cancers 2020, 12, 3016. [Google Scholar] [CrossRef]
- Goicoechea, S.M.; Awadia, S.; Garcia-Mata, R. I’m coming to GEF you: Regulation of RhoGEFs during cell migration. Cell Adhes. Migr. 2014, 8, 535–549. [Google Scholar] [CrossRef]
- Garrett, T.A.; Van Buul, J.D.; Burridge, K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell Res. 2007, 313, 3285–3297. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Brekken, R.A.; Vander Kooi, C.W.; Mercurio, A.M. P-Rex1 Promotes Resistance to VEGF/VEGFR-Targeted Therapy in Prostate Cancer. Cell Rep. 2016, 14, 2193–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Bryant, D.M.; Mostov, K.E. Molecular Regulation of Lumen Morphogenesis. Curr. Biol. 2011, 21, R126–R136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzinger, D.B.; Mariani, L.; Weis, J.; Woyke, T.; Berens, T.J.; McDonough, W.; Sloan, A.; Coons, S.W.; Berens, M.E. Gene Expression Profile of Glioblastoma Multiforme Invasive Phenotype Points to New Therapeutic Targets. Neoplasia 2005, 7, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, A.L.M.; Lun, X.; Rahn, J.J.; Liacini, A.; Wang, L.; Hamilton, M.G.; Parney, I.F.; Hempstead, B.L.; Robbins, S.M.; Forsyth, P.A.; et al. The p75 Neurotrophin Receptor Is a Central Regulator of Glioma Invasion. PLoS Biol. 2007, 5, e212. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.L.; Pokorny, J.L.; Schroeder, M.A.; Sarkaria, J.N. Establishment, Maintenance, and In Vitro and In Vivo Applications of Primary Human Glioblastoma Multiforme (GBM) Xenograft Models for Translational Biology Studies and Drug Discovery. In Current Protocols in Pharmacology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2011; Chapter 14, Unit 14.16. [Google Scholar]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar] [CrossRef]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Staruschenko, A.; Sorokin, A. Role of βpix in the kidney. Front. Physiol. 2012, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Li, X.; Premont, R.T. Expanding functions of GIT Arf GTPase-activating proteins, PIX Rho guanine nucleotide exchange factors and GIT–PIX complexes. J. Cell Sci. 2016, 129, 1963–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Baird, D.; Peng, X.; Wang, J.; Ly, T.; Guan, J.-L.; Cerione, R.A. Cool-1 functions as an essential regulatory node for EGFreceptor- and Src-mediated cell growth. Nat. Cell Biol. 2006, 8, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Han, X.; Hsiao, C.; Yates III, J.R.; Waterman, C.M. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat. Cell Biol. 2011, 13, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López Tobón, A.; Suresh, M.; Jin, J.; Vitriolo, A.; Pietralla, T.; Tedford, K.; Bossenz, M.; Mahnken, K.; Kiefer, F.; Testa, G.; et al. The guanine nucleotide exchange factor Arhgef7/βPix promotes axon formation upstream of TC10. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Omelchenko, T.; Rabadan, M.A.; Hernáandez-Martínez, R.; Grego-Bessa, J.; Anderson, K.V.; Hall, A. Β-Pix Directs Collective Migration of Anterior Visceral Endoderm Cells in the Early Mouse Embryo. Genes Dev. 2014, 28, 2764–2777. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.M.; Folts, C.J.; Cui, W.; Bardin, A.L.; Walter, K.; Carson-Walter, E.; Vescovi, A.; Noble, M. Cool-1-Mediated Inhibition of c-Cbl Modulates Multiple Critical Properties of Glioblastomas, Including the Ability to Generate Tumors In Vivo. Stem Cells 2014, 32, 1124–1135. [Google Scholar] [CrossRef]
- Lei, X.; Deng, L.; Liu, D.; Liao, S.; Dai, H.; Li, J.; Rong, J.; Wang, Z.; Huang, G.; Tang, C.; et al. ARHGEF7 promotes metastasis of colorectal adenocarcinoma by regulating the motility of cancer cells. Int. J. Oncol. 2018, 53, 1980–1996. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.-H.; Huang, W.-C.; Chang, J.-W.; Chang, K.-J.; Kuo, W.-H.; Wang, M.-Y.; Lin, K.-Y.; Uen, Y.-H.; Hou, M.-F.; Lin, C.-M.; et al. MicroRNA-149 targets GIT1 to suppress integrin signaling and breast cancer metastasis. Oncogene 2014, 33, 4496–4507. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Tsunoda, T.; Riku, M.; Inaguma, S.; Inoko, A.; Murakami, H.; Ikeda, H.; Matsuda, M.; Kasai, K. Indispensable role of STIL in the regulation of cancer cell motility through the lamellipodial accumulation of ARHGEF7–PAK1 complex. Oncogene 2020, 39, 1931–1943. [Google Scholar] [CrossRef]
- Ngok, S.P.; Anastasiadis, P.Z. Rho GEFs in endothelial junctions. Tissue Barriers 2013, 1, e27132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Malyukova, I.; Mikaelyan, A.; Fu, P.; Birukov, K.G. Tiam1 and βPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J. Cell. Physiol. 2007, 211, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; Connor, K.; Sweeney, K.; Miller, I.S.; O’Farrell, A.; Ruiz-Hernandez, E.; Murray, D.M.; Duffy, G.P.; Wolfe, A.; Mastrobattista, E.; et al. RGD-decorated cholesterol stabilized polyplexes for targeted siRNA delivery to glioblastoma cells. Drug Deliv. Transl. Res. 2019, 9, 679–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Han, I.; Wu, L.; Zeng, X. Targeted Nanotechnology in Glioblastoma Multiforme. Front. Pharm. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zottel, A.; Videtič Paska, A.; Jovčevska, I. Nanotechnology Meets Oncology: Nanomaterials in Brain Cancer Research, Diagnosis and Therapy. Materials 2019, 12, 1588. [Google Scholar] [CrossRef] [Green Version]
- Kislin, K.L.; McDonough, W.S.; Eschbacher, J.M.; Armstrong, B.A.; Berens, M.E. NHERF-1: Modulator of Glioblastoma Cell Migration and Invasion. Neoplasia 2009, 11, 377-IN7. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017, 19, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef]
- Piccinini, F.; Tesei, A.; Arienti, C.; Bevilacqua, A. Cell Counting and Viability Assessment of 2D and 3D Cell Cultures: Expected Reliability of the Trypan Blue Assay. Biol. Proced. Online 2017, 19, 8. [Google Scholar] [CrossRef]
- Jarzabek, M.A.; Huszthy, P.C.; Skaftnesmo, K.O.; McCormack, E.; Dicker, P.; Prehn, J.H.M.; Bjerkvig, R.; Byrne, A.T. In vivo bioluminescence imaging validation of a human biopsy-derived orthotopic mouse model of glioblastoma multiforme. Mol. Imaging 2013, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, K.J.; Jarzabek, M.A.; Dicker, P.; O’Brien, D.F.; Callanan, J.J.; Byrne, A.T.; Prehn, J.H.M. Validation of an imageable surgical resection animal model of Glioblastoma (GBM). J. Neurosci. Methods 2014, 233, 99–104. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Connor, K.; Murray, D.W.; Jarzabek, M.A.; Tran, N.L.; White, K.; Dicker, P.; Sweeney, K.J.; O’Halloran, P.J.; MacCarthy, B.; Shiels, L.P.; et al. Targeting the RhoGEF βPIX/COOL-1 in Glioblastoma: Proof of Concept Studies. Cancers 2020, 12, 3531. https://doi.org/10.3390/cancers12123531
Connor K, Murray DW, Jarzabek MA, Tran NL, White K, Dicker P, Sweeney KJ, O’Halloran PJ, MacCarthy B, Shiels LP, et al. Targeting the RhoGEF βPIX/COOL-1 in Glioblastoma: Proof of Concept Studies. Cancers. 2020; 12(12):3531. https://doi.org/10.3390/cancers12123531
Chicago/Turabian StyleConnor, Kate, David W. Murray, Monika A. Jarzabek, Nhan L. Tran, Kieron White, Patrick Dicker, Kieron J. Sweeney, Philip J. O’Halloran, Brian MacCarthy, Liam P. Shiels, and et al. 2020. "Targeting the RhoGEF βPIX/COOL-1 in Glioblastoma: Proof of Concept Studies" Cancers 12, no. 12: 3531. https://doi.org/10.3390/cancers12123531