
Balancing the CD38 Expression on Effector and Target Cells in Daratumumab-Mediated NK Cell ADCC against Multiple Myeloma


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. CRISPR Plasmids
2.3. Lentiviral Vector Production and Cell Transduction
2.4. Staining by Flow Cytometry
2.5. Quantification of CD38 Expression
2.6. Irradiation of Effector Cells
2.7. PBMCs and NK Cells Isolation
2.8. Incubation of Target Cells with Adjuvants
2.9. Incubation of Effector Cells with Anti-CD38 Nanobody or Anti-NKG2A Antibody
2.10. Cellular Cytotoxicity Assays: Flow Cytometry
2.11. Cellular Cytotoxicity Assays: Calcein Release Assay
2.12. Statistical Analyses
3. Results
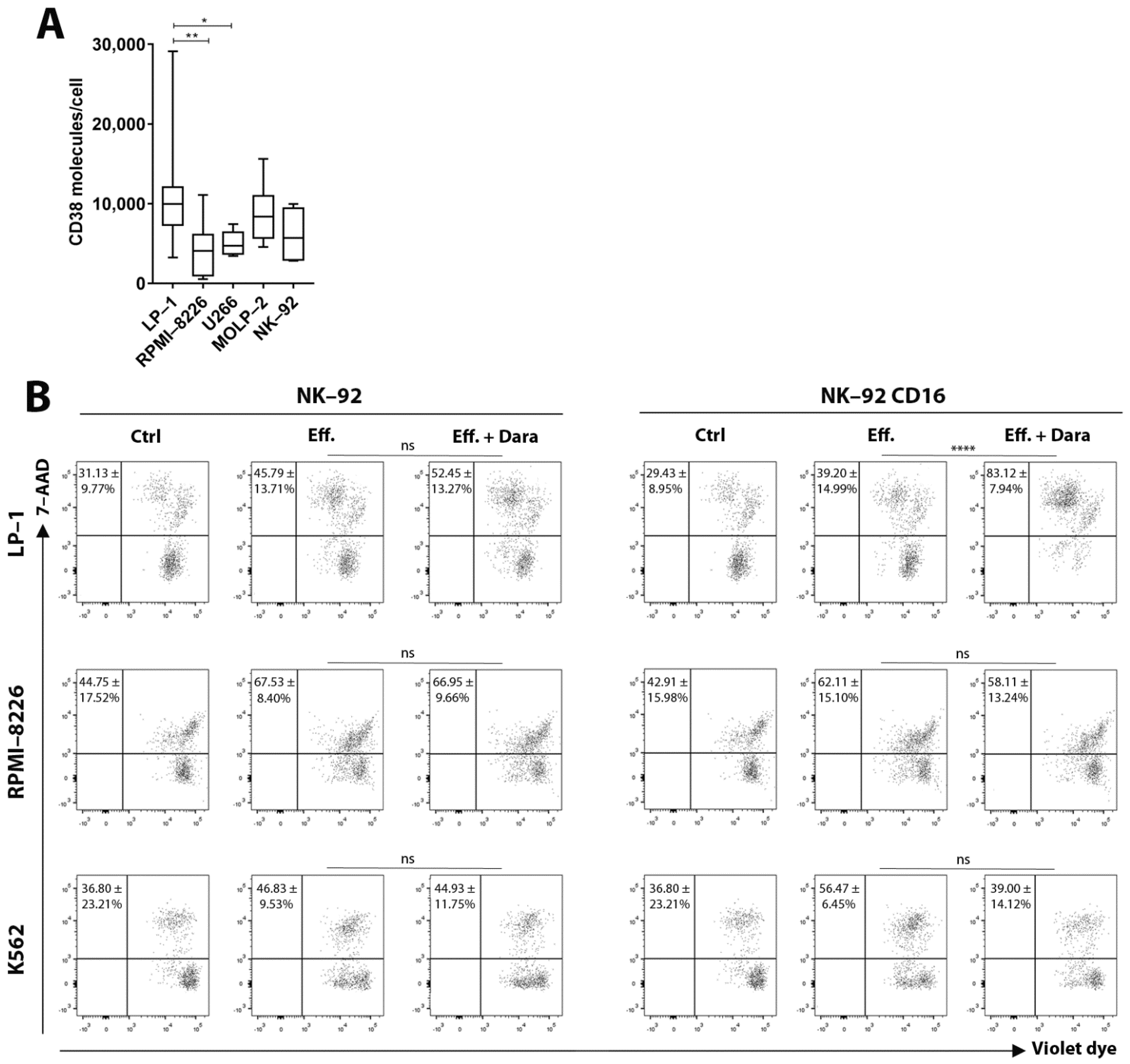
3.1. ADCC—Dependent Tumor Cell Cytotoxicity: Flow Cytometry
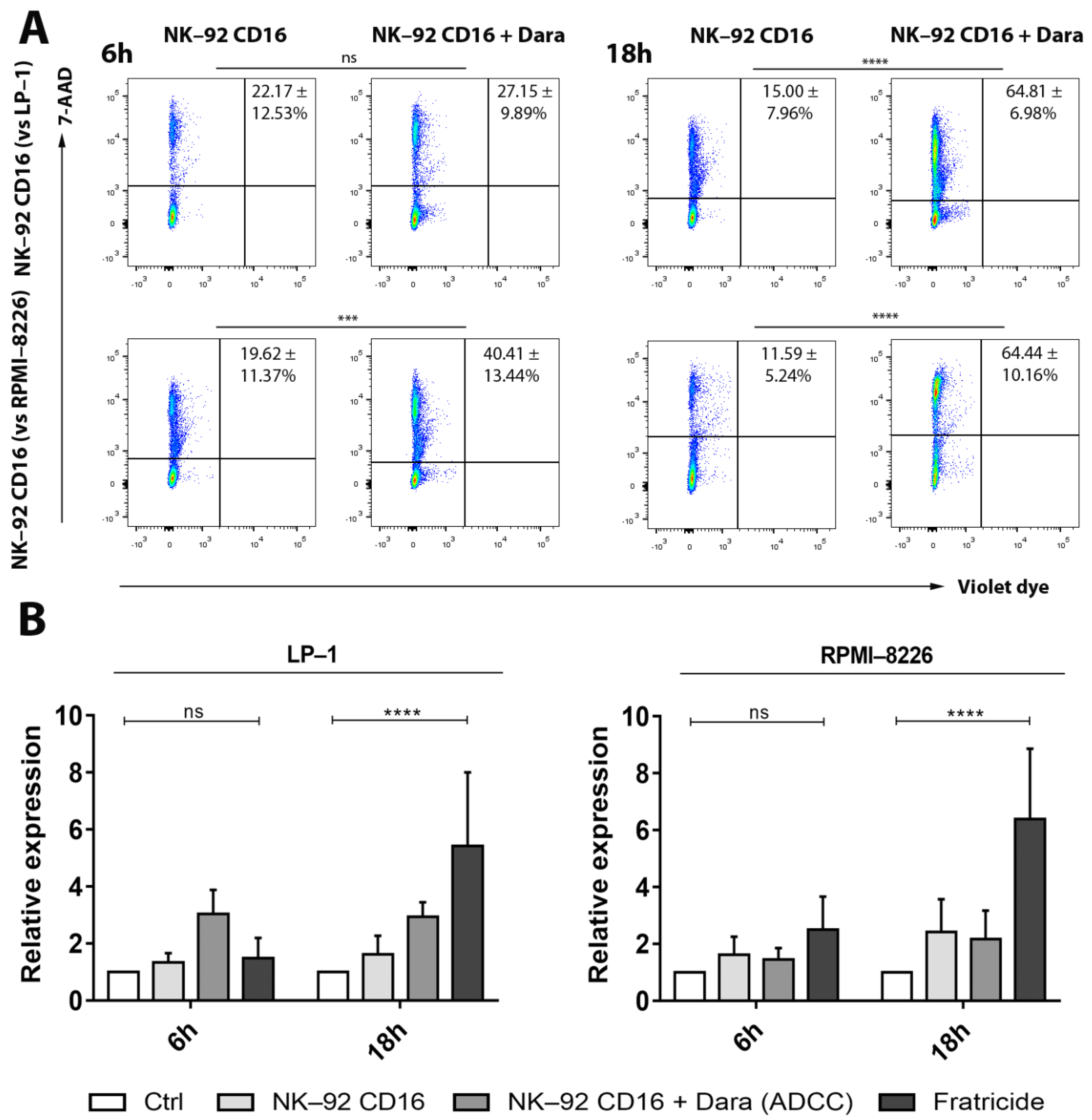
3.2. Fratricide between NK-92 CD16a Effector Cells: Flow Cytometry
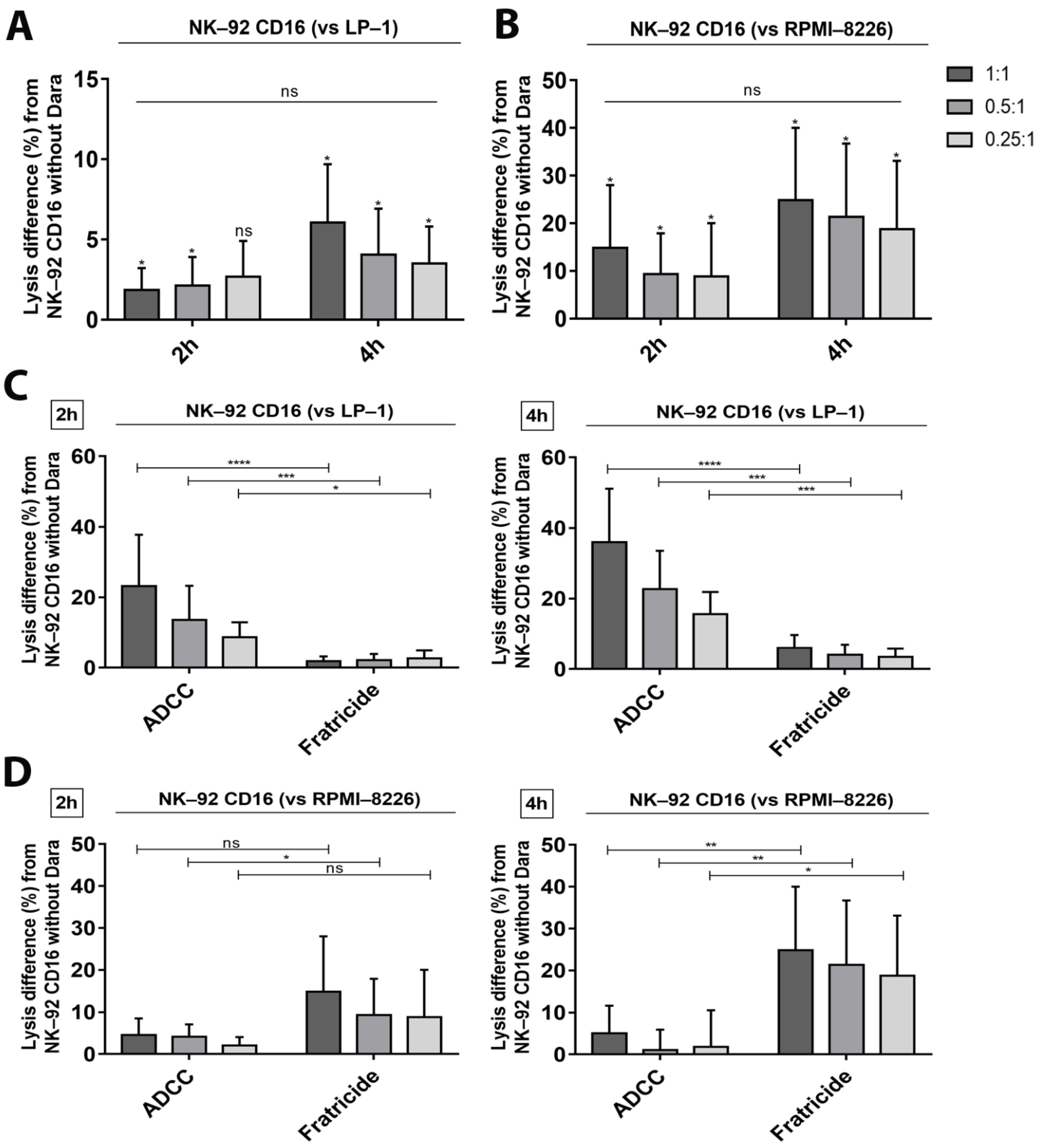
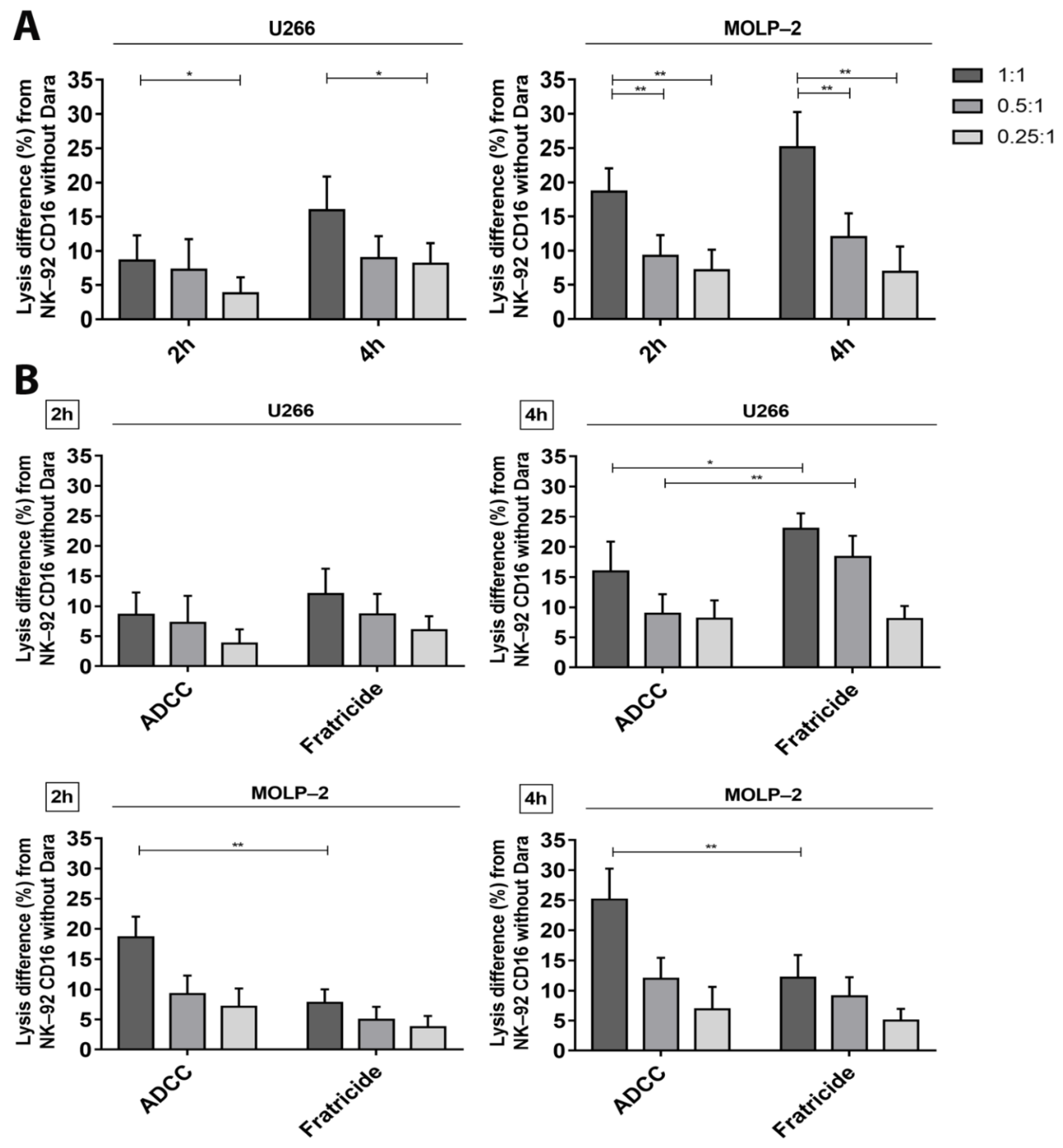
3.3. Confirmation of Cytotoxicity and Fratricide by the Calcein Release Assay
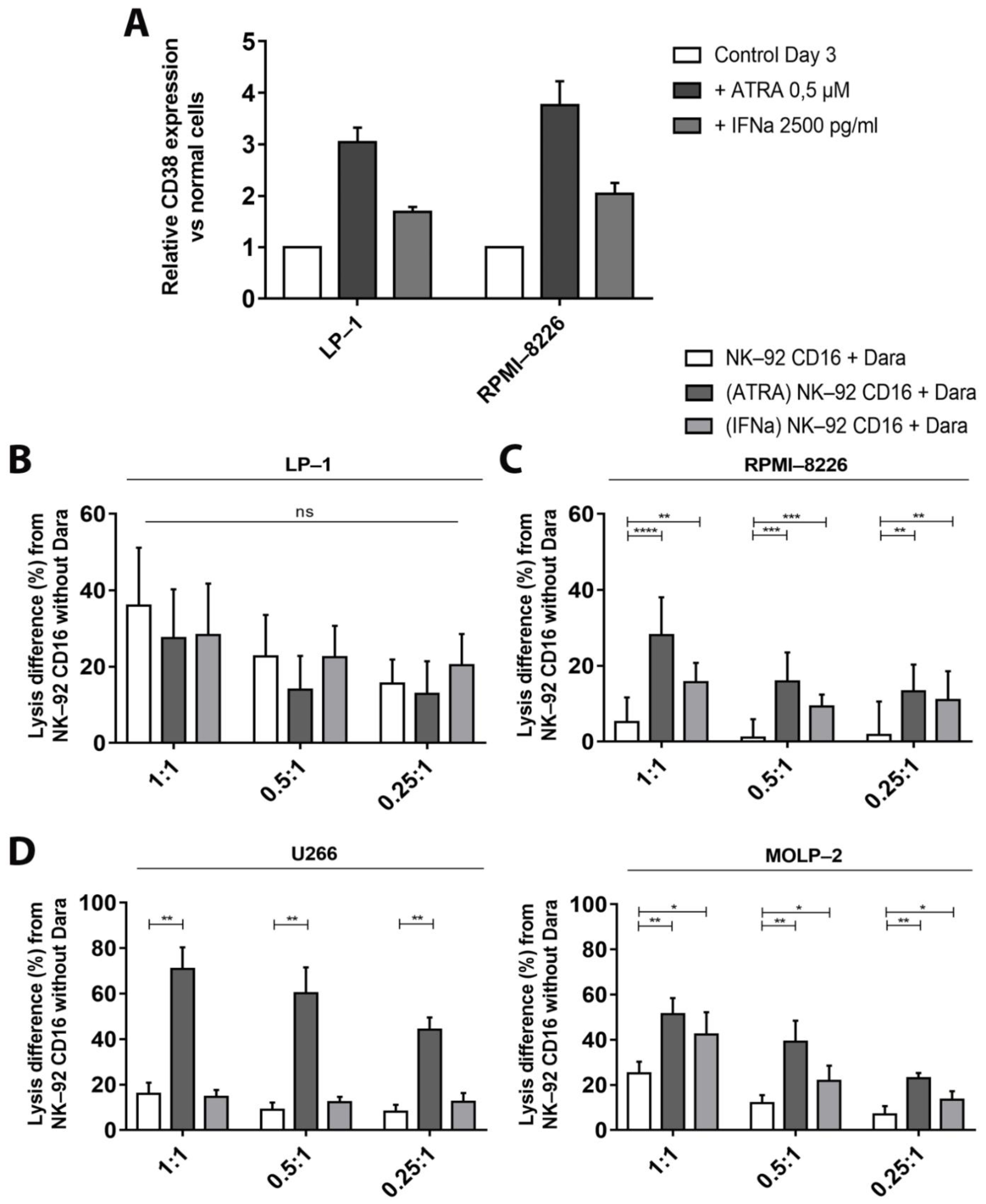
3.4. Increase of CD38 Expression after Adding of Adjuvants
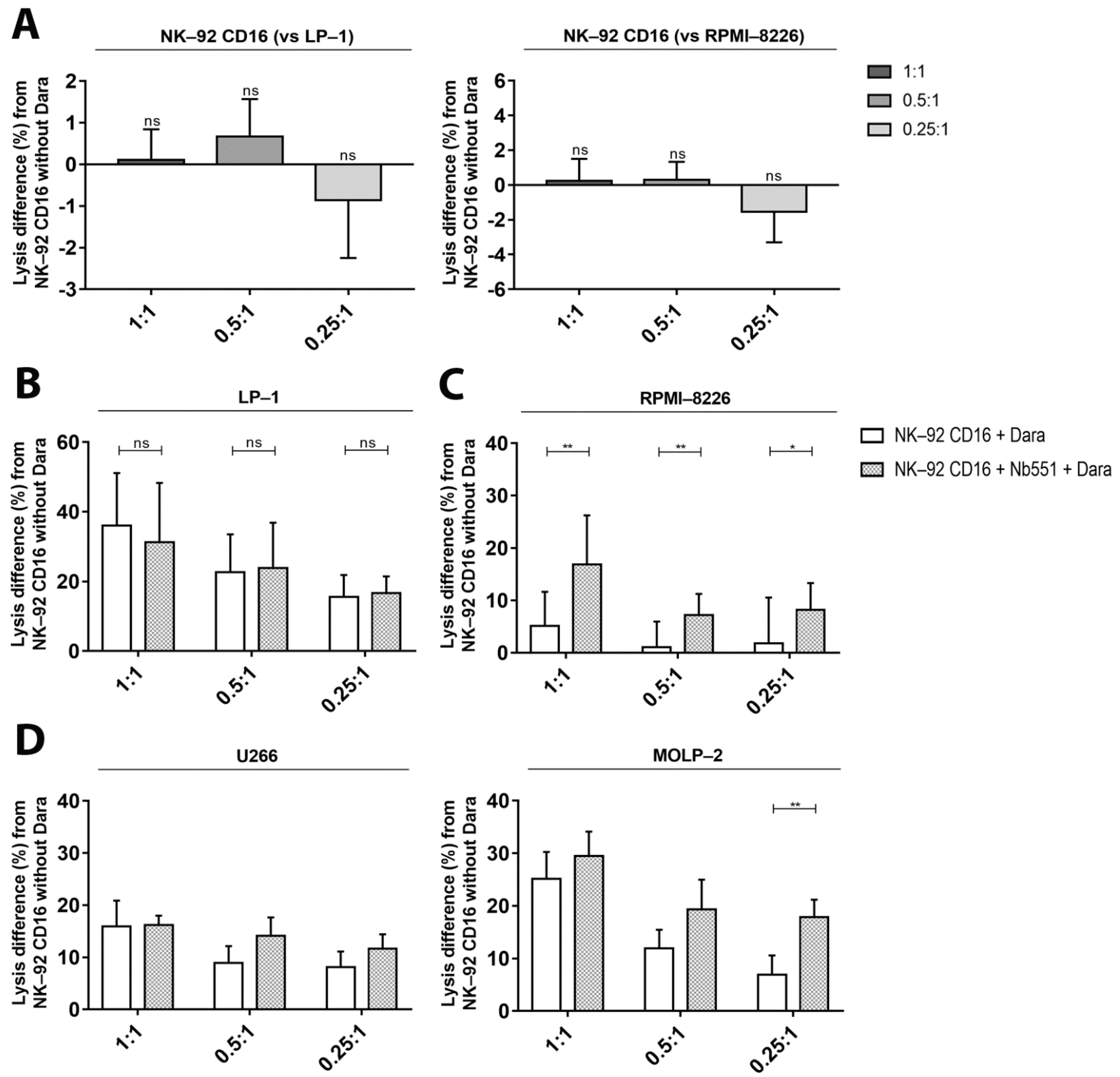
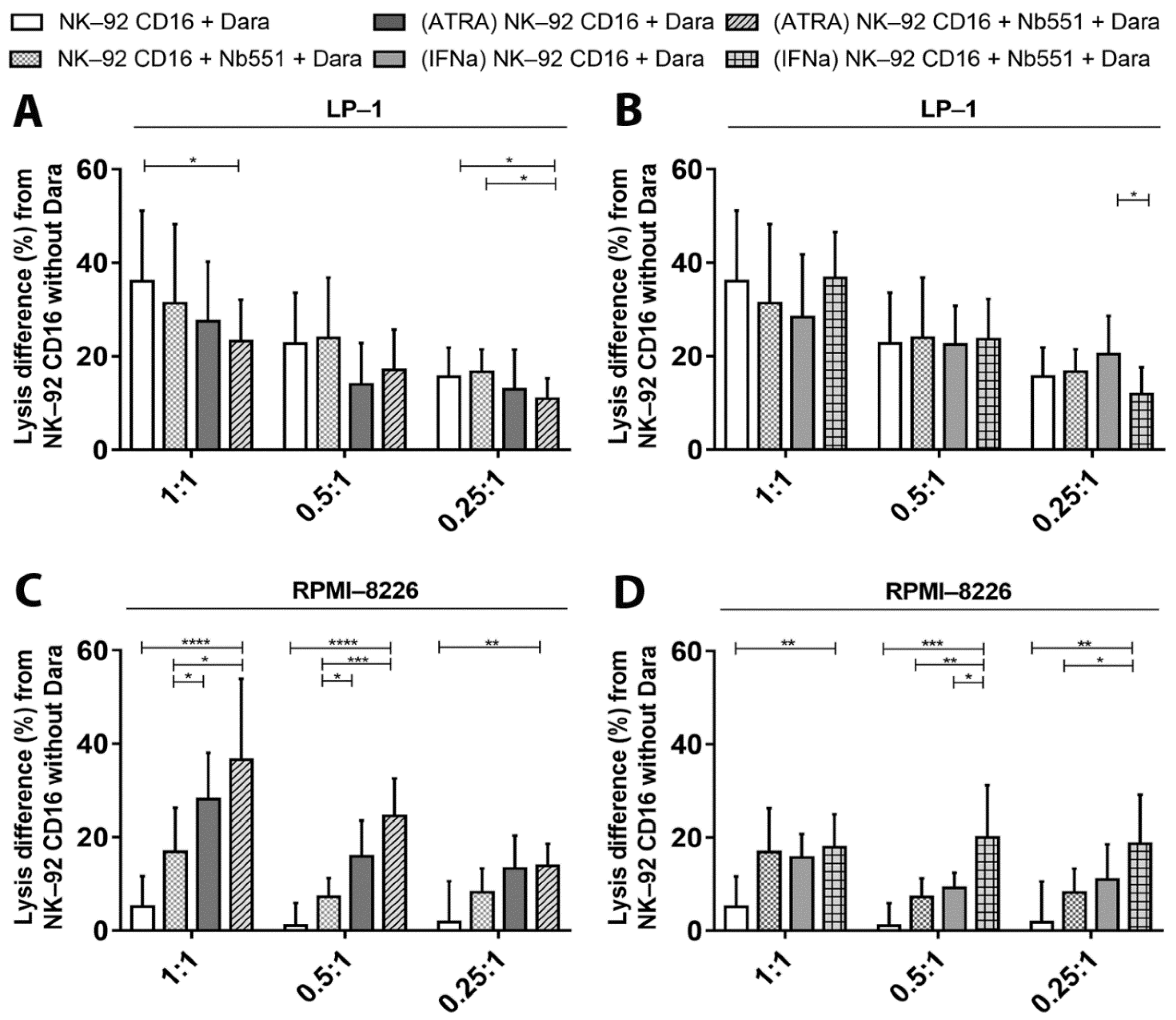
3.5. ADCC—Dependent Tumor Cell Cytotoxicity after Increase of CD38 Expression and Fratricide Inhibition
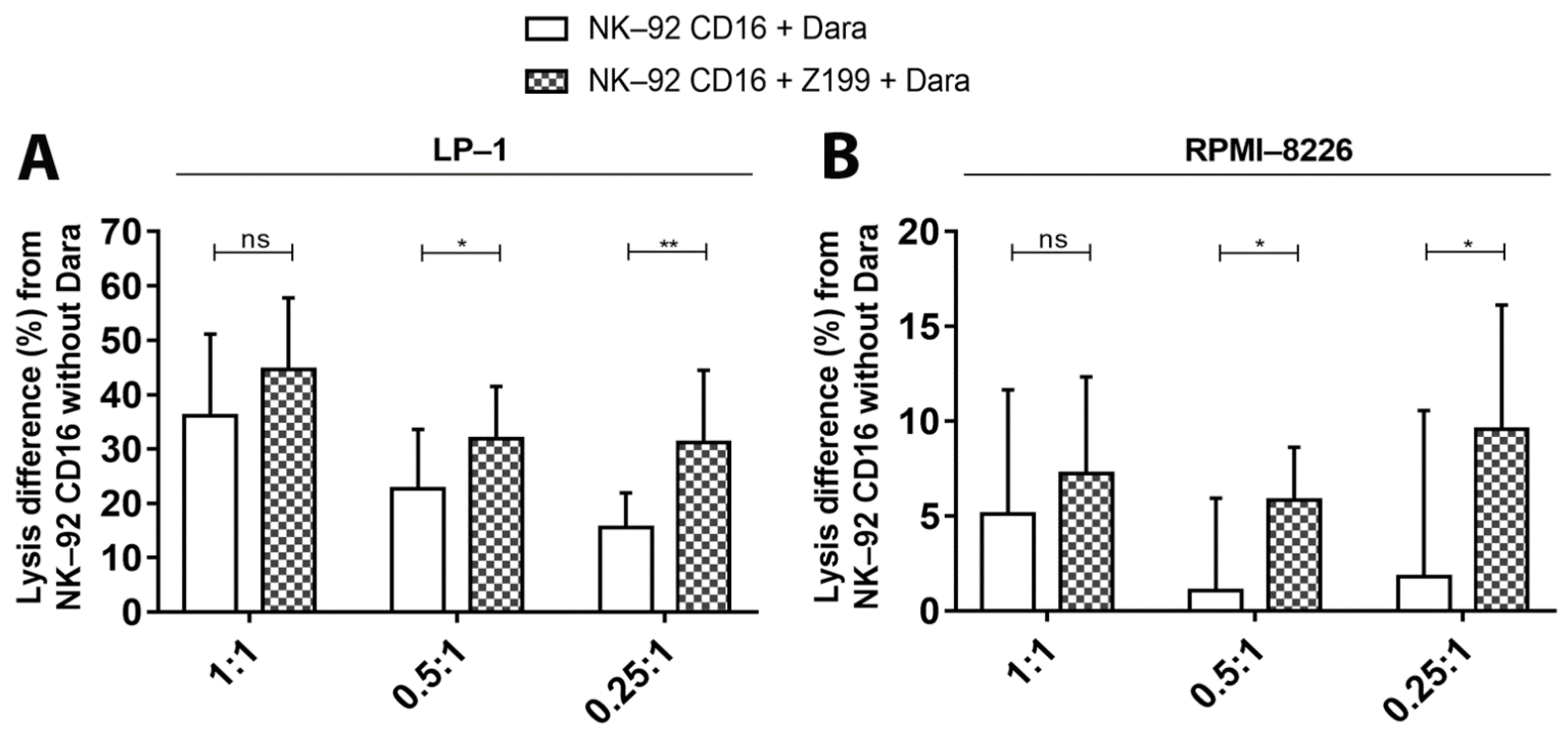
3.6. Inhibition of Inhibitor Receptor NKG2A on NK-92 CD16a Cells with a mAb (CD159a)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, S.-F.; Lin, L.; Xing, L.; Yu, T.; Wen, K.; Anderson, K.C.; Tai, Y.-T. Monoclonal Antibody: A New Treatment Strategy against Multiple Myeloma. Antibodies 2017, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Abramson, H.N. Monoclonal Antibodies for the Treatment of Multiple Myeloma: An Update. Int. J. Mol. Sci. 2018, 19, 3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fast, L.D.; Hansen, J.A.; Newman, W. Evidence for T cell nature and heterogeneity within natural killer (NK) and antibody-dependent cellular cytotoxicity (ADCC) effectors: A comparison with cytolytic T lymphocytes (CTL). J. Immunol. 1981, 127, 448–452. [Google Scholar] [PubMed]
- Mahaweni, N.M.; Bos, G.M.J.; Mitsiades, C.S.; Tilanus, M.G.J.; Wieten, L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol. Immunother. 2018, 67, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubach, J.P.; Tai, Y.-T.; Richardson, P.G.; Anderson, K.C. Daratumumab granted breakthrough drug status. Expert Opin. Investig. Drugs 2014, 23, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk. Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Wong, S.W.; Comenzo, R.L. CD38 Monoclonal Antibody Therapies for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2015, 15, 635–645. [Google Scholar] [CrossRef]
- McKeage, K. Daratumumab: First Global Approval. Drugs 2016, 76, 275–281. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; Van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; Van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Casneuf, T.; Xu, X.S.; Adams, H.C.; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo–Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [Green Version]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.; Law, A.D.; Routy, B.; Denhollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clémenceau, B.; Vivien, R.; Pellat, C.; Foss, M.; Thibault, G.; Vié, H. The human natural killer cytotoxic cell line NK-92, once armed with a murine CD16 receptor, represents a convenient cellular tool for the screening of mouse mAbs according to their ADCC potential. MAbs 2013, 5, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emi, N.; Friedmann, T.; Yee, J.K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J. Virol. 1991, 65, 1202–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Qi, S.; Unger, M.; Hou, Y.N.; Deng, Q.W.; Liu, J.; Lam, C.M.C.; Wang, X.W.; Xin, D.; Zhang, P.; et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci. Rep. 2016, 6, 27055. [Google Scholar] [CrossRef] [Green Version]
- Nijhof, I.S.; Groen, R.W.J.; Lokhorst, H.M.; Van Kessel, B.; Bloem, A.C.; Van Velzen, J.; De Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, S.; Eulenburg, C.Z.; Hildebrandt, Y.; Stubig, T.; Sierich, H.; Badbaran, A.; Eiermann, T.H.; Binder, T.M.C.; Kroger, N. Impact of the NK cell receptor LIR-1 (ILT-2/CD85j/LILRB1) on cytotoxicity against multiple myeloma. Clin. Dev. Immunol. 2012, 2012, 652130. [Google Scholar] [CrossRef] [PubMed]
- Kararoudi, M.N.; Nagai, Y.; Elmas, E.; Pereira, M.D.S.F.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]
- Sarkar, S.; Chauhan, S.K.S.; Daly, J.; Natoni, A.; Fairfield, H.; Henderson, R.; Nolan, E.; Swan, D.; Hu, J.; Reagan, M.R.; et al. The CD38low natural killer cell line KHYG1 transiently expressing CD16F158V in combination with daratumumab targets multiple myeloma cells with minimal effector NK cell fratricide. Cancer Immunol. Immunother. 2020, 69, 421–434. [Google Scholar] [CrossRef]
- Lewandowski, D.; Linassier, C.; Iochmann, S.; Degenne, M.; Domenech, J.; Colombat, P.; Binet, C.; Herault, O. Phosphatidylinositol 3-kinases are involved in the all-trans retinoic acid-induced upregulation of CD38 antigen on human haematopoietic cells. Br. J. Haematol. 2002, 118, 535–544. [Google Scholar] [CrossRef]
- Mihara, K.; Yoshida, T.; Ishida, S.; Takei, Y.; Kitanaka, A.; Shimoda, K.; Morishita, K.; Takihara, Y.; Ichinohe, T. All-trans retinoic acid and interferon-α increase CD38 expression on adult T-cell leukemia cells and sensitize them to T cells bearing anti-CD38 chimeric antigen receptors. Blood Cancer J. 2016, 6, e421. [Google Scholar] [CrossRef] [Green Version]
- García-Guerrero, E.; Götz, R.; Doose, S.; Sauer, M.; Rodríguez-Gil, A.; Nerreter, T.; Kortüm, K.M.; Pérez-Simón, J.A.; Einsele, H.; Hudecek, M.; et al. Upregulation of CD38 expression on multiple myeloma cells by novel HDAC6 inhibitors is a class effect and augments the efficacy of daratumumab. Leukemia 2021, 35, 201–214. [Google Scholar] [CrossRef]
- Fatholahi, M.; Valencia, M.; Mark, A.; Bi, M.; Syed, S.; Zhang, Y.; Taura, T.; Yun, Y.; Wilson, D.; Chattopadhyay, N.; et al. TAK-573, an anti-CD38-targeted attenuated interferon alpha (IFNα) fusion protein, showed anti-myeloma tumor responses in combination with standard of care (SOC) agents in multiple myeloma (MM) xenograft tumor models in vivo. Clin. Lymphoma Myeloma Leuk. 2019, 19, e116. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Battella, S.; De Federicis, D.; Galandrini, R.; Palmieri, G. Harnessing CD16-Mediated NK Cell Functions to Enhance Therapeutic Efficacy of Tumor-Targeting mAbs. Cancers 2021, 13, 2500. [Google Scholar] [CrossRef] [PubMed]
- Motais, B.; Charvátová, S.; Walek, Z.; Hrdinka, M.; Smolarczyk, R.; Cichoń, T.; Czapla, J.; Giebel, S.; Šimíček, M.; Jelínek, T.; et al. Selection, Expansion, and Unique Pretreatment of Allogeneic Human Natural Killer Cells with Anti-CD38 Monoclonal Antibody for Efficient Multiple Myeloma Treatment. Cells 2021, 10, 967. [Google Scholar] [CrossRef] [PubMed]
- Reina-Ortiz, C.; Constantinides, M.; Fayd-Herbe-De-Maudave, A.; Présumey, J.; Hernandez, J.; Cartron, G.; Giraldos, D.; Díez, R.; Izquierdo, I.; Azaceta, G.; et al. Expanded NK cells from umbilical cord blood and adult peripheral blood combined with daratumumab are effective against tumor cells from multiple myeloma patients. OncoImmunology 2020, 10, 1853314. [Google Scholar] [CrossRef]
- Adams, H.C., 3rd; Stevenaert, F.; Krejcik, J.; van der Borght, K.; Smets, T.; Bald, J.; Abraham, Y.; Ceulemans, H.; Chiu, C.; Vanhoof, G.; et al. High-Parameter Mass Cytometry Evaluation of Relapsed/Refractory Multiple Myeloma Patients Treated with Daratumumab Demonstrates Immune Modulation as a Novel Mechanism of Action. Cytometry A 2019, 95, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; Van De Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejeune, M.; Duray, E.; Peipp, M.; Clémenceau, B.; Baron, F.; Beguin, Y.; Caers, J. Balancing the CD38 Expression on Effector and Target Cells in Daratumumab-Mediated NK Cell ADCC against Multiple Myeloma. Cancers 2021, 13, 3072. https://doi.org/10.3390/cancers13123072
Lejeune M, Duray E, Peipp M, Clémenceau B, Baron F, Beguin Y, Caers J. Balancing the CD38 Expression on Effector and Target Cells in Daratumumab-Mediated NK Cell ADCC against Multiple Myeloma. Cancers. 2021; 13(12):3072. https://doi.org/10.3390/cancers13123072
Chicago/Turabian StyleLejeune, Margaux, Elodie Duray, Matthias Peipp, Béatrice Clémenceau, Frédéric Baron, Yves Beguin, and Jo Caers. 2021. "Balancing the CD38 Expression on Effector and Target Cells in Daratumumab-Mediated NK Cell ADCC against Multiple Myeloma" Cancers 13, no. 12: 3072. https://doi.org/10.3390/cancers13123072
APA StyleLejeune, M., Duray, E., Peipp, M., Clémenceau, B., Baron, F., Beguin, Y., & Caers, J. (2021). Balancing the CD38 Expression on Effector and Target Cells in Daratumumab-Mediated NK Cell ADCC against Multiple Myeloma. Cancers, 13(12), 3072. https://doi.org/10.3390/cancers13123072