
Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors



Abstract
:Simple Summary
Abstract
1. Introduction
2. Small-Molecule Inhibitors Targeting the UPS
2.1. Proteasome Inhibitors
2.2. Targeting E1 and E2 Enzymes
2.3. Targeting E3 Ligases
2.3.1. RING-Type E3 Ligases
Targeting the MDM2–p53 Interaction
CRL/SCF RING E3 Ligases
Other RING E3s
2.3.2. HECT-Type E3 Ligase Inhibitors
2.3.3. RBR-Type E3 Ligase Inhibitors
2.4. Chemical Inhibitors of DUBs
2.4.1. USP7 Inhibitors
2.4.2. Other DUB Inhibitors
3. Targeted Protein Degradation
3.1. PROTACs
3.1.1. First Generation PROTACs
3.1.2. Development of Small-Molecule PROTACs
3.1.3. PROTAC Variations
3.1.4. PROTAC Future Directions
3.2. Other Notable Approaches to Targeted Protein Modulation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uckelmann, M.; Sixma, T.K. Histone ubiquitination in the DNA damage response. DNA Repair 2017, 56, 92–101. [Google Scholar] [CrossRef]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- Bednash, J.S.; Mallampalli, R.K. Regulation of inflammasomes by ubiquitination. Cell. Mol. Immunol. 2016, 13, 722–728. [Google Scholar] [CrossRef]
- Leestemaker, Y.; Ovaa, H. Tools to investigate the ubiquitin proteasome system. Drug Discov. Today Technol. 2017, 26, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, Hypoxia, and Cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passmore, L.A.; Barford, D. Getting into position: The catalytic mechanisms of protein ubiquitylation. Biochem. J. 2004, 379, 513–525. [Google Scholar] [CrossRef]
- Li, W.; Ye, Y. Polyubiquitin chains: Functions, structures, and mechanisms. Cell. Mol. Life Sci. 2008, 65, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Ronai, Z.A. Monoubiquitination in proteasomal degradation. Proc. Natl. Acad. Sci. USA 2016, 113, 8894–8896. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N. Ubiquitin System. Int. J. Mol. Sci. 2018, 19, 1080. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Urbé, S.; Komander, D. Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 2019, 20, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, 3–9. [Google Scholar] [CrossRef]
- Mofers, A.; Pellegrini, P.; Linder, S.; D’Arcy, P. Proteasome-associated deubiquitinases and cancer. Cancer Metastasis Rev. 2017, 36, 635–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauwers, E.; Jacob, C.; André, B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol. 2009, 185, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell. Mol. Biol. Lett. 2021, 26, 1–17. [Google Scholar] [CrossRef]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Linder, S.; Bazzaro, M. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers. Cancers 2020, 12, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Li, S.; Shu, H.-B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [Green Version]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Clague, M.; Urbé, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.; Otín, C.L. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2012, 31, 2373–2388. [Google Scholar] [CrossRef] [Green Version]
- Coleman, K.E.; Huang, T.T. In a Class of Its Own: A New Family of Deubiquitinases Promotes Genome Stability. Mol. Cell 2018, 70, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Mao, J.-H.; Kim, I.-J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 Targets mTOR for Degradation and Cooperates with PTEN in Tumor Suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Biol. 2011, 23, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Kimura, Y.; Tokuda, M.; Honda, S.; Hirose, S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006, 7, 1019–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavie, J.; De Belvalet, H.; Sonon, S.; Ion, A.M.; Dumon, E.; Melser, S.; Lacombe, D.; Dupuy, J.-W.; Lalou, C.; Bénard, G. Ubiquitin-Dependent Degradation of Mitochondrial Proteins Regulates Energy Metabolism. Cell Rep. 2018, 23, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol. Cell. Proteom. 2011, 10, R110-006924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 Regulates an NF-κB-Independent Cell-Death Switch in TNF Signaling. Curr. Biol. 2007, 17, 418–424. [Google Scholar] [CrossRef] [Green Version]
- Bazzaro, M.; Lee, M.K.; Zoso, A.; Stirling, W.L.; Santillan, A.; Shih, I.-M.; Roden, R.B. Ubiquitin-Proteasome System Stress Sensitizes Ovarian Cancer to Proteasome Inhibitor–Induced Apoptosis. Cancer Res. 2006, 66, 3754–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Hideshima, T.; Anderson, K.C. Bortezomib (PS-341): A Novel, First-in-Class Proteasome Inhibitor for the Treatment of Multiple Myeloma and Other Cancers. Cancer Control 2003, 10, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Veggiani, G.; Gerpe, M.C.R.; Sidhu, S.S.; Zhang, W. Emerging drug development technologies targeting ubiquitination for cancer therapeutics. Pharmacol. Ther. 2019, 199, 139–154. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Crews, C.M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [Google Scholar] [CrossRef] [Green Version]
- Hoeller, D.; Dikic, I. Targeting the ubiquitin system in cancer therapy. Nat. Cell Biol. 2009, 458, 438–444. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.A.; Stewart, A.K.; Vallone, M.; Molineaux, C.J.; Kunkel, L.A.; Gerecitano, J.F.; Orlowski, R. A Phase 1 Dose Escalation Study of the Safety and Pharmacokinetics of the Novel Proteasome Inhibitor Carfilzomib (PR-171) in Patients with Hematologic Malignancies. Clin. Cancer Res. 2009, 15, 7085–7091. [Google Scholar] [CrossRef] [Green Version]
- Demo, S.D.; Kirk, C.J.; Aujay, M.A.; Buchholz, T.J.; Dajee, M.; Ho, M.N.; Jiang, J.; Laidig, G.J.; Lewis, E.R.; Parlati, F.; et al. Antitumor Activity of PR-171, a Novel Irreversible Inhibitor of the Proteasome. Cancer Res. 2007, 67, 6383–6391. [Google Scholar] [CrossRef] [Green Version]
- Rajan, A.M.; Kumar, S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016, 6, e451. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Baz, R.; Wang, M.; Jakubowiak, A.J.; Laubach, J.P.; Harvey, R.D.; Talpaz, M.; Berg, D.; Liu, G.; Yu, J.; et al. Phase 1 study of twice-weekly ixazomib, an oral proteasome inhibitor, in relapsed/refractory multiple myeloma patients. Blood 2014, 124, 1038–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP. Nat. Cell Biol. 2010, 467, 179–184. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.-K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef]
- Coughlin, K.; Anchoori, R.; Iizuka, Y.; Meints, J.; MacNeill, L.; Vogel, R.; Orlowski, R.; Lee, M.K.; Roden, R.B.; Bazzaro, M. Small-Molecule RA-9 Inhibits Proteasome-Associated DUBs and Ovarian Cancer In Vitro and In Vivo via Exacerbating Unfolded Protein Responses. Clin. Cancer Res. 2014, 20, 3174–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anchoori, R.K.; Karanam, B.; Peng, S.; Wang, J.W.; Jiang, R.; Tanno, T.; Orlowski, R.; Matsui, W.; Zhao, M.; Rudek, M.A.; et al. A bis-Benzylidine Piperidone Targeting Proteasome Ubiquitin Receptor RPN13/ADRM1 as a Therapy for Cancer. Cancer Cell 2013, 24, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Cao, Y.; Greten, F.; Li, Z.-W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-κB as a Therapeutic Target in Multiple Myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [Green Version]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.; Irwin, D.; Stadtmauer, E.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Bancroft, C.C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-κB, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Baiz, D.; Pozzato, G.; Dapas, B.; Farra, R.; Scaggiante, B.; Grassi, M.; Uxa, L.; Giansante, C.; Zennaro, C.; Guarnieri, G.; et al. Bortezomib arrests the proliferation of hepatocellular carcinoma cells HepG2 and JHH6 by differentially affecting E2F1, p21 and p27 levels. Biochimie 2009, 91, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kontopodis, E.; Kotsakis, A.; Kentepozidis, N.; Syrigos, K.; Ziras, N.; Moutsos, M.; Filippa, G.; Mala, A.; Vamvakas, L.; Mavroudis, D.; et al. A phase II, open-label trial of bortezomib (VELCADE®) in combination with gemcitabine and cisplatin in patients with locally advanced or metastatic non-small cell lung cancer. Cancer Chemother. Pharmacol. 2016, 77, 949–956. [Google Scholar] [CrossRef]
- Fisher, R.I.; Bernstein, S.H.; Kahl, B.S.; Djulbegovic, B.; Robertson, M.J.; De Vos, S.; Epner, E.; Krishnan, A.; Leonard, J.P.; Lonial, S.; et al. Multicenter Phase II Study of Bortezomib in Patients with Relapsed or Refractory Mantle Cell Lymphoma. J. Clin. Oncol. 2006, 24, 4867–4874. [Google Scholar] [CrossRef] [Green Version]
- Cavaletti, G.; Jakubowiak, A.J. Peripheral neuropathy during bortezomib treatment of multiple myeloma: A review of recent studies. Leuk. Lymphoma 2010, 51, 1178–1187. [Google Scholar] [CrossRef]
- Shin, Y.K.; Jang, S.Y.; Lee, H.K.; Jung, J.; Suh, D.J.; Seo, S.-Y.; Park, H.T. Pathological adaptive responses of Schwann cells to endoplasmic reticulum stress in bortezomib-induced peripheral neuropathy. Glia 2010, 58, 1961–1976. [Google Scholar] [CrossRef]
- Suzuki, E.; Demo, S.; Deu, E.; Keats, J.; Arastu-Kapur, S.; Bergsagel, P.L.; Bennett, M.K.; Kirk, C.J. Molecular Mechanisms of Bortezomib Resistant Adenocarcinoma Cells. PLoS ONE 2011, 6, e27996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; Van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Q.; Baker, D.; Ovaa, H. Small molecules that target the ubiquitin system. Biochem. Soc. Trans. 2020, 48, 479–497. [Google Scholar] [CrossRef] [Green Version]
- Girnius, S.K.; Lee, S.; Kambhampati, S.; Rose, M.G.; Mohiuddin, A.; Houranieh, A.; Zimelman, A.; Grady, T.; Mehta, P.; Behler, C.; et al. A Phase II trial of weekly bortezomib and dexamethasone in veterans with newly diagnosed multiple myeloma not eligible for or who deferred autologous stem cell transplantation. Br. J. Haematol. 2015, 169, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Straka, C.; Knop, S.; Vogel, M.; Müller, J.; Kropff, M.; Metzner, B.; Langer, C.; Sayer, H.; Jung, W.; Dürk, H.A.; et al. Bortezomib consolidation following autologous transplant in younger and older patients with newly diagnosed multiple myeloma in two phase III trials. Eur. J. Haematol. 2019, 103, 255–267. [Google Scholar] [CrossRef]
- Moreau, P.; Pylypenko, H.; Grosicki, S.; Karamanesht, I.; Leleu, X.; Grishunina, M.; Rekhtman, G.; Masliak, Z.; Robak, T.; Shubina, A.; et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: A randomised, phase 3, non-inferiority study. Lancet Oncol. 2011, 12, 431–440. [Google Scholar] [CrossRef]
- Berenson, J.R.; Hilger, J.D.; Yellin, O.; Dichmann, R.; Pateldonnelly, D.; Boccia, R.V.; Bessudo, A.; Stampleman, L.; Gravenor, D.S.; Eshaghian, S.; et al. Replacement of bortezomib with carfilzomib for multiple myeloma patients progressing from bortezomib combination therapy. Leukemia 2014, 28, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Joshua, D.; Chng, W.-J.; Palumbo, A.; Goldschmidt, H.; Hajek, R.; Facon, T.; Ludwig, H.; Pour, L.; Niesvizky, R.; et al. Impact of prior treatment on patients with relapsed multiple myeloma treated with carfilzomib and dexamethasone vs bortezomib and dexamethasone in the phase 3 ENDEAVOR study. Leukemia 2016, 31, 115–122. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Gregory, T.K.; Faber, E.A.; Hart, L.L.; Mace, J.R.; Arrowsmith, E.R.; Flinn, I.W.; Matous, J.V. A phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed or relapsed/refractory multiple myeloma: Final analysis of second dose-expansion cohort. Am. J. Hematol. 2021, 96, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, P.; Paba-Prada, C.E.; Voorhees, P.M.; Frye, J.; Chang, Y.-L.; Moreau, P.; Zonder, J.; Boccia, R.; Shain, K.H. Efficacy and safety results from a phase 1b/2, multicenter, open-label study of oprozomib and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Leuk. Res. 2019, 83, 106172. [Google Scholar] [CrossRef]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the Proteasome Inhibitor MLN9708 in Preclinical Models of Human Cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and In Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.C.; Fitzgerald, M.; Bannerman, B.; Donelan, J.; Bano, K.; Terkelsen, J.; Bradley, D.P.; Subakan, O.; Silva, M.D.; Liu, R.; et al. Antitumor Activity of the Investigational Proteasome Inhibitor MLN9708 in Mouse Models of B-cell and Plasma Cell Malignancies. Clin. Cancer Res. 2011, 17, 7313–7323. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, H.; Poenisch, W.; Knop, S.; Egle, A.; Schreder, M.; Lechner, D.; Hajek, R.; Gunsilius, E.; Krenosz, K.J.; Petzer, A.; et al. Ixazomib–Thalidomide–Dexamethasone for induction therapy followed by Ixazomib maintenance treatment in patients with relapsed/refractory multiple myeloma. Br. J. Cancer 2019, 121, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Yang, H.; Hanley, M.J.; Zhang, S.; Liu, R.; Kumar, S.; Richardson, P.G.; Skacel, T.; Venkatakrishnan, K. Dose and Schedule Selection of the Oral Proteasome Inhibitor Ixazomib in Relapsed/Refractory Multiple Myeloma: Clinical and Model-Based Analyses. Target. Oncol. 2017, 12, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jiang, Y.; Ding, S.; Li, J.; Song, N.; Ren, Y.; Hong, D.; Wu, C.; Li, B.; Wang, F.; et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018, 28, 1186–1194. [Google Scholar] [CrossRef]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef]
- Zhang, X.; Pellegrini, P.; Saei, A.A.; Hillert, E.-K.; Mazurkiewicz, M.; Olofsson, M.H.; Zubarev, R.; D’Arcy, P.; Linder, S. The deubiquitinase inhibitor b-AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem. Pharmacol. 2018, 156, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Bartholomeusz, G.; Talpaz, M.; Donato, N.J. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood 2011, 117, 3151–3162. [Google Scholar] [CrossRef] [Green Version]
- Potu, H.; Kandarpa, M.; Peterson, L.F.; Durham, A.; Donato, N.J.; Talpaz, M. Downregulation of SOX2 by inhibition of Usp9X induces apoptosis in melanoma. Oncotarget 2021, 12, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.V.; Tamayo, A.T.; Li, C.; Bornmann, W.; Priebe, W.; Ford, R.J. Degrasyn Potentiates the Antitumor Effects of Bortezomib in Mantle Cell Lymphoma Cells In vitro and In vivo: Therapeutic Implications. Mol. Cancer Ther. 2010, 9, 2026–2036. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Umezawa, Y.; Ishida, S.; Okada, K.; Nogami, A.; Miura, O. Inhibition of USP9X induces apoptosis in FLT3-ITD-positive AML cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. 2019, 453, 84–94. [Google Scholar] [CrossRef]
- Li, J.; Yakushi, T.; Parlati, F.; MacKinnon, A.L.; Perez, C.; Ma, Y.; Carter, K.P.; Colayco, S.; Magnuson, G.; Brown, B.; et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn. Nat. Chem. Biol. 2017, 13, 486–493. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Y.; Santos, B.D.S.S.D.; Wang, F.; Ma, Y.; Perez, C.; Yang, Y.; Peng, J.; Cohen, S.M.; Chou, T.-F.; et al. Epidithiodiketopiperazines Inhibit Protein Degradation by Targeting Proteasome Deubiquitinase Rpn. Cell Chem. Biol. 2018, 25, 1350–1358.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nat. Cell Biol. 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Milhollen, M.A.; Traore, T.; Adams-Duffy, J.; Thomas, M.P.; Berger, A.J.; Dang, L.; Dick, L.R.; Garnsey, J.J.; Koenig, E.; Langston, S.P.; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-κB–dependent lymphoma. Blood 2010, 116, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; Dwyer, M.O.; Nawrocki, S.T.; et al. Brief report Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Sedarati, F.; Faller, D.V.; Zhao, D.; Faessel, H.M.; Chowdhury, S.; Bolleddula, J.; Li, Y.; Venkatakrishnan, K.; Papai, Z. Phase I study assessing the mass balance, pharmacokinetics, and excretion of [14C]-pevonedistat, a NEDD8-activating enzyme inhibitor in patients with advanced solid tumors. Investig. New Drugs 2021, 39, 488–498. [Google Scholar] [CrossRef]
- Faessel, H.M.; Mould, D.R.; Zhou, X.; Faller, U.V.; Sedarati, F.; Venkatakrishnan, K. Population pharmacokinetics of pevonedistat alone or in combination with standard of care in patients with solid tumours or haematological malignancies. Br. J. Clin. Pharmacol. 2019, 85, 2568–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockhart, A.C.; Bauer, T.M.; Aggarwal, C.; Lee, C.B.; Harvey, R.D.; Cohen, R.B.; Sedarati, F.; Nip, T.K.; Faessel, H.; Dash, A.B.; et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Investig. New Drugs 2019, 37, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.; Cruz, J.; Erba, H.P.; Berdeja, J.G.; Tam, W.; Vardhanabhuti, S.; et al. Pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, combined with azacitidine in patients with AML. Blood 2018, 131, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, D.; Tang, X.; Pelletier, B.; Orlicky, S.; Xie, W.; Plantevin, V.; Neculai, D.; Chou, Y.-C.; Ogunjimi, A.; Al-Hakim, A.; et al. An Allosteric Inhibitor of the Human Cdc34 Ubiquitin-Conjugating Enzyme. Cell 2011, 145, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Pulvino, M.; Liang, Y.; Oleksyn, D.; DeRan, M.; Van Pelt, E.; Shapiro, J.; Sanz, I.; Chen, L.; Zhao, J. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012, 120, 1668–1677. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472–9481. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Xu, D.D.; Zhang, D.; Chen, J.; Gao, F.G. PYR-41 and Thalidomide Impair Dendritic Cell Cross-Presentation by Inhibiting Myddosome Formation and Attenuating the Endosomal Recruitments of p97 and Sec61 via NF-κB Inactivation. J. Immunol. Res. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, G.S. Role of Ubiquitin Ligases and the Proteasome in Oncogenesis: Novel Targets for Anticancer Therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Sarantopoulos, J.; Shapiro, G.I.; Cohen, R.B.; Clark, J.W.; Kauh, J.S.; Weiss, G.J.; Cleary, J.M.; Mahalingam, D.; Pickard, M.D.; Faessel, H.M.; et al. Phase I Study of the Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (TAK-924/MLN4924) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Lovering, R.; Hanson, I.M.; Borden, K.L.; Martin, S.; O’Reilly, N.; Evan, G.I.; Rahman, D.; Pappin, D.; Trowsdale, J.; Freemont, P. Identification and preliminary characterization of a protein motif related to the zinc finger. Proc. Natl. Acad. Sci. USA 1993, 90, 2112–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.B.; Pruneda, J.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhou, P. DCAFs, the Missing Link of the CUL4-DDB1 Ubiquitin Ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef]
- Schreiber, A.; Stengel, F.; Zhang, Z.; Enchev, R.I.; Kong, E.H.; Morris, E.; Robinson, C.V.; Da Fonseca, P.C.A.; Barford, D. Structural basis for the subunit assembly of the anaphase-promoting complex. Nat. Cell Biol. 2011, 470, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. Erratum: HECT E3 ubiquitin ligases—Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchiero, P.; Barbarotto, E.; Tiribelli, M.; Zerbinati, C.; Di Iasio, M.G.; Gonelli, A.; Cavazzini, F.; Campioni, D.; Fanin, R.; Cuneo, A.; et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood 2006, 107, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Melloni, E.; Di Iasio, M.G.; Tiribelli, M.; Rimondi, E.; Corallini, F.; Gattei, V.; Zauli, G. Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood 2009, 113, 4300–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM. Leukemia 2008, 22, 730–739. [Google Scholar] [CrossRef] [Green Version]
- Kaindl, U.; Morak, M.; Portsmouth, C.; Mecklenbräuker, A.; Kauer, M.; Zeginigg, M.; Attarbaschi, A.; Haas, O.A.; Panzer-Grümayer, R. Blocking ETV6/RUNX1-induced MDM2 overexpression by Nutlin-3 reactivates p53 signaling in childhood leukemia. Leukemia 2013, 28, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Tonsing-Carter, A.; Bailey, B.J.; Saadatzadeh, M.R.; Ding, J.; Wang, H.; Sinn, A.L.; Peterman, K.M.; Spragins, T.K.; Silver, J.M.; Sprouse, A.A.; et al. Potentiation of Carboplatin-Mediated DNA Damage by the Mdm2 Modulator Nutlin-3a in a Humanized Orthotopic Breast-to-Lung Metastatic Model. Mol. Cancer Ther. 2015, 14, 2850–2863. [Google Scholar] [CrossRef] [Green Version]
- Tosoni, D.; Pambianco, S.; Soppo, B.E.; Zecchini, S.; Bertalot, G.; Pruneri, G.; Viale, G.; Di Fiore, P.P.; Pece, S. Pre-clinical validation of a selective anti-cancer stem cell therapy for Numb-deficient human breast cancers. EMBO Mol. Med. 2017, 9, 655–671. [Google Scholar] [CrossRef]
- Sarisozen, C.; Tan, Y.; Liu, J.; Bilir, C.; Shen, L.; Filipczak, N.; Porter, T.M.; Torchilin, V.P. MDM2 antagonist-loaded targeted micelles in combination with doxorubicin: Effective synergism against human glioblastoma via p53 re-activation. J. Drug Target. 2019, 27, 624–633. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, A.; Tolcher, A.; Beeram, M.; Nemunaitis, J.; Weiss, G.J.; Bhalla, K.; Agrawal, M.; Nichols, G.; Middleton, S.; Beryozkina, A.; et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 587–595. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.W.; Assouline, S.; Strair, R.K.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.; Lu, M.; Kosiorek, H.; Virtgaym, E.; Xia, L.; Sandy, L.; Mesa, R.; Petersen, B.; Farnoud, N.; Najfeld, V.; et al. Oral idasanutlin in patients with polycythemia vera. Blood 2019, 134, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, K.; Papayannidis, C.; Vey, N.; Dickinson, M.J.; Kelly, K.R.; Assouline, S.; Kasner, M.; Seiter, K.; Drummond, M.W.; Yoon, S.-S.; et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study⋆. Leuk. Res. 2021, 100, 106489. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Futur. Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An Optimized Inhibitor of MDM2–p53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Macé, S.; Tuffal, G.; et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef]
- De Weger, V.A.; De Jonge, M.; Langenberg, M.H.G.; Schellens, J.H.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Gojo, I.; Patnaik, M.M.; Minden, M.D.; Kantarjian, H.; Johnson-Levonas, A.O.; Fancourt, C.; Lam, R.; Jones, M.B.; Knox, C.D.; et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk. Res. 2016, 48, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.J.; Banerji, U.; Mahipal, A.; Somaiah, N.; Hirsch, H.; Fancourt, C.; Johnson-Levonas, A.O.; Lam, R.; Meister, A.K.; Russo, G.; et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 1304–1311. [Google Scholar] [CrossRef]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a Potent, Selective, and Orally Bioavailable MDM2–p53 Inhibitor in Clinical Development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Werner, L.R.; Huang, S.; Francis, D.M.; Armstrong, E.A.; Ma, F.; Li, C.; Iyer, G.; Canon, J.; Harari, P.M. Small Molecule Inhibition of MDM2–p53 Interaction Augments Radiation Response in Human Tumors. Mol. Cancer Ther. 2015, 14, 1994–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef]
- Gluck, W.L.; Gounder, M.M.; Frank, R.; Eskens, F.; Blay, J.Y.; Cassier, P.A.; Soria, J.-C.; Chawla, S.; De Weger, V.; Wagner, A.J.; et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Investig. New Drugs 2019, 38, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wünschel, J.; Toedling, J.; Deubzer, H.E.; Künkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2017, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Gounder, M.M.; Weise, A.M.; Schwartz, G.K.; Carvajal, R.D.; Kumar, P.; Zernovak, O.; Beck, A.; Doyle, J.; Mendell-Harary, J.; et al. A phase 1 study of MDM2 inhibitor DS-3032b in patients with well/de-differentiated liposarcoma (WD/DD LPS), solid tumors (ST) and lymphomas (L). J. Clin. Oncol. 2018, 36, 11514. [Google Scholar] [CrossRef]
- Dinardo, C.D.; Rosenthal, M.J.; Andreeff, M.; Zernovak, O.; Kumar, P.; Gajee, R.; Chen, S.; Rosen, M.; Song, S.; Kochan, J.; et al. Phase 1 Dose Escalation Study of MDM2 Inhibitor DS-3032b in Patients with Hematological Malignancies—Preliminary Results. Blood 2016, 128, 593. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [Green Version]
- Holzer, P.; Chène, P.; Ferretti, S.; Furet, P.; Gabriel, T.; Gruenenfelder, B.; Guagnano, V.; Hofmann, F.; Kallen, J.; Mah, R.; et al. Abstract 4855: Discovery of NVP-HDM201—First disclosure of a Next-Generation Mdm2 inhibitor with superior characteristics. Cancer Chem. 2016, 76, 4855. [Google Scholar] [CrossRef]
- Ferretti, S.; Rebmann, R.; Berger, M.; Santacroce, F.; Albrecht, G.; Pollehn, K.; Sterker, D.; Wartmann, M.; Hueber, A.; Wiesmann, M.; et al. Abstract 1224: Insights into the mechanism of action of NVP-HDM201, a differentiated and versatile Next-Generation small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. Exp. Mol. Ther. 2016, 76, 1224. [Google Scholar] [CrossRef]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B.; et al. Discovery of 4-((3′R,4′S,5′R)-6″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2″-oxodispiro[cyclohexane-1,2′-pyrrolidine-3′,3″-indoline]-5′-carboxamido)bicyclo[2.2.2]octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Lakhani, N.J.; Li, Y.; Men, L.; Wang, H.; Ji, J.; Tang, Y.; Liang, Z.; Amaya, A.; Estkowski, K.; et al. A phase I study of a novel MDM2 antagonist APG-115 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3126. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Karim, R.; Tang, Y.; Wang, H.; Ji, J.; Chen, J.; Kaiser, A.; Sheladia, P.; Ahmad, M.; Liang, Z.; et al. Phase Ib study of a novel, small-molecule MDM2 inhibitor APG-115 combined with pembrolizumab in U.S. patients with metastatic solid tumors. J. Clin. Oncol. 2020, 38, 3512. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maïga, S.; Lodé, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; le Gouill, S.; Amiot, M.; et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneberg, S.; Cherif, H.; Lazarevic, V.; Andersson, P.-O.; Von Euler, M.; Juliusson, G.; Lehmann, S. An open-label phase I dose-finding study of APR-246 in hematological malignancies. Blood Cancer J. 2016, 6, e447. [Google Scholar] [CrossRef] [Green Version]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-Based Design of Spiro-oxindoles as Potent, Specific Small-Molecule Inhibitors of the MDM2−p53 Interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [Green Version]
- Sasiela, C.A.; Stewart, D.H.; Kitagaki, J.; Safiran, Y.J.; Yang, Y.; Weissman, A.M.; Oberoi, P.; Davydov, I.V.; Goncharova, E.; Beutler, J.A.; et al. Identification of Inhibitors for MDM2 Ubiquitin Ligase Activity from Natural Product Extracts by a Novel High-Throughput Electrochemiluminescent Screen. J. Biomol. Screen. 2008, 13, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-D. Cytotoxic effect of a non-peptidic small molecular inhibitor of the p53-HDM2 interaction on tumor cells. World J. Gastroenterol. 2005, 11, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, J.; Agama, K.K.; Pommier, Y.; Yang, Y.; Weissman, A.M. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol. Cancer Ther. 2008, 7, 2445–2454. [Google Scholar] [CrossRef] [Green Version]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 Ligase Inhibitors Using a Cell-Based Ubiquitination Assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ma, X.; Ren, S.; Buolamwini, J.K.; Yan, C. A Small-Molecule Inhibitor of MDMX Activates p53 and Induces Apoptosis. Mol. Cancer Ther. 2011, 10, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-L.; Weng, H.-Y.; Wang, L.-Q.; Yu, C.-H.; Huang, Q.-J.; Zhao, P.-P.; Wen, J.-Z.; Zhou, H.; Qu, L.-H. Triggering Fbw7-Mediated Proteasomal Degradation of c-Myc by Oridonin Induces Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2012, 11, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Orlicky, S.; Tang, X.; Neduva, V.; Elowe, N.; Brown, E.D.; Sicheri, F.; Tyers, M. An allosteric inhibitor of substrate recognition by the SCFCdc4 ubiquitin ligase. Nat. Biotechnol. 2010, 28, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xie, W.; Kuhn, D.J.; Voorhees, P.M.; Lopez-Girona, A.; Mendy, D.; Corral, L.G.; Krenitsky, V.P.; Xu, W.; Parseval, L.M.-D.; et al. Targeting the p27 E3 ligase SCFSkp2 results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008, 111, 4690–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico-Bautista, E.; Yang, C.-C.; Lu, L.; Roth, G.P.; A Wolf, D. Chemical genetics approach to restoring p27Kip1 reveals novel compounds with antiproliferative activity in prostate cancer cells. BMC Biol. 2010, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Grigoryan, A.V.; Li, Y.; Hao, B.; Pagano, M.; Cardozo, T.J. Specific Small Molecule Inhibitors of Skp2-Mediated p27 Degradation. Chem. Biol. 2012, 19, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-H.; Morrow, J.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.-J.; Bai, H.-Y.; Li, Z.-H.; Zou, J.; Chen, J.-W.; Zheng, F.; Zhang, J.-X.; Mai, S.-J.; Zeng, M.-S.; Sun, H.-D.; et al. Longikaurin A, a natural ent-kaurane, induces G2/M phase arrest via downregulation of Skp2 and apoptosis induction through ROS/JNK/c-Jun pathway in hepatocellular carcinoma cells. Cell Death Dis. 2014, 5, e1137. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Lin, C.-L.; Lin, J.-K. 1,2,3,4,6-Penta-O-galloyl-β-d-glucose, Quercetin, Curcumin and Lycopene Induce Cell-Cycle Arrest in MDA-MB-231 and BT474 Cells through Downregulation of Skp2 Protein. J. Agric. Food Chem. 2011, 59, 6765–6775. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Zhou, X.; Wang, L.; Yin, X.; Wang, Z. Curcumin inhibits cell growth and invasion and induces apoptosis through down-regulation of Skp2 in pancreatic cancer cells. Am. J. Cancer Res. 2016, 6, 1949–1962. [Google Scholar] [PubMed]
- Wang, L.; Ye, X.; Cai, X.; Su, J.; Ma, R.; Yin, X.; Zhou, X.; Li, H.; Wang, Z. Curcumin suppresses cell growth and invasion and induces apoptosis by down-regulation of Skp2 pathway in glioma cells. Oncotarget 2015, 6, 18027–18037. [Google Scholar] [CrossRef] [Green Version]
- James, M.I.; Iwuji, C.; Irving, G.; Karmokar, A.; Higgins, J.A.; Griffin-Teal, N.; Thomas, A.; Greaves, P.; Cai, H.; Patel, S.R.; et al. Curcumin inhibits cancer stem cell phenotypes in ex vivo models of colorectal liver metastases, and is clinically safe and tolerable in combination with FOLFOX chemotherapy. Cancer Lett. 2015, 364, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howells, L.M.; Iwuji, C.O.O.; Irving, G.R.B.; Barber, S.; Walter, H.; Sidat, Z.; Griffin-Teall, N.; Singh, R.; Foreman, N.; Patel, S.R.; et al. Curcumin Combined with FOLFOX Chemotherapy Is Safe and Tolerable in Patients with Metastatic Colorectal Cancer in a Randomized Phase IIa Trial. J. Nutr. 2019, 149, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Blees, J.S.; Bokesch, H.R.; Rübsamen, D.; Schulz, K.; Milke, L.; Bajer, M.M.; Gustafson, K.R.; Henrich, C.J.; McMahon, J.B.; Colburn, N.H.; et al. Erioflorin Stabilizes the Tumor Suppressor Pdcd4 by Inhibiting Its Interaction with the E3-ligase β-TrCP1. PLoS ONE 2012, 7, e46567. [Google Scholar] [CrossRef]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.-S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef]
- Flygare, J.A.; Beresini, M.; Budha, N.; Chan, H.; Chan, I.T.; Cheeti, S.; Cohen, F.; Deshayes, K.; Doerner, K.; Eckhardt, S.G.; et al. Discovery of a Potent Small-Molecule Antagonist of Inhibitor of Apoptosis (IAP) Proteins and Clinical Candidate for the Treatment of Cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Kondapuram, S.K.; Yang, T.-H.; Syed, S.B.; Cheng, S.M.; Lin, T.-Y.; Lin, Y.-C.; Coumar, M.S.; Chang, J.-Y.; Leung, E.; et al. The SMAC mimetic LCL161 is a direct ABCB1/MDR1-ATPase activity modulator and BIRC5/Survivin expression down-regulator in cancer cells. Toxicol. Appl. Pharmacol. 2020, 401, 115080. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Wilson, G.S.; Lie, S.; Wu, G.; Hu, Z.; Hebbard, L.; Duan, W.; George, J.; Qiao, L. Synergistic effects of IAP inhibitor LCL161 and paclitaxel on hepatocellular carcinoma cells. Cancer Lett. 2014, 351, 232–241. [Google Scholar] [CrossRef]
- Shekhar, T.M.; Burvenich, I.J.G.; Harris, M.A.; Rigopoulos, A.; Zanker, D.; Spurling, A.; Parker, B.S.; Walkley, C.R.; Scott, A.M.; Hawkins, C.J. Smac mimetics LCL161 and GDC-0152 inhibit osteosarcoma growth and metastasis in mice. BMC Cancer 2019, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I Dose-Escalation Study of LCL161, an Oral Inhibitor of Apoptosis Proteins Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef]
- Cai, Q.; Sun, H.; Peng, Y.; Lu, J.; Nikolovska-Coleska, Z.; McEachern, D.; Liu, L.; Qiu, S.; Yang, C.-Y.; Miller, R.; et al. A Potent and Orally Active Antagonist (SM-406/AT-406) of Multiple Inhibitor of Apoptosis Proteins (IAPs) in Clinical Development for Cancer Treatment. J. Med. Chem. 2011, 54, 2714–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Fujibuchi, W.; Ishida, K.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; Shibata, C.; et al. Inhibitor of apoptosis protein family as diagnostic markers and therapeutic targets of colorectal cancer. Surg. Today 2011, 41, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Brunckhorst, M.K.; Lerner, D.; Wang, S.; Yu, Q. AT-406, an orally active antagonist of multiple inhibitor of apoptosis proteins, inhibits progression of human ovarian cancer. Cancer Biol. Ther. 2012, 13, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.I.; Smith, D.C.; Pitot, H.C.; Brill, J.M.; Chugh, R.; Rouits, E.; Rubin, J.; Strickler, J.; Vuagniaux, G.; Sorensen, J.M.; et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: Results of a first-in-man study. Cancer Chemother. Pharmacol. 2015, 75, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-S.; Tao, Y.; Le Tourneau, C.; Pointreau, Y.; Sire, C.; Kaminsky, M.-C.; Coutte, A.; Alfonsi, M.; Boisselier, P.; Martin, L.; et al. Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: A double-blind, multicentre, randomised, phase 2 study. Lancet Oncol. 2020, 21, 1173–1187. [Google Scholar] [CrossRef]
- Katragadda, L.; Carter, B.Z.; Borthakur, G. XIAP antisense therapy with AEG 35156 in acute myeloid leukemia. Expert Opin. Investig. Drugs 2013, 22, 663–670. [Google Scholar] [CrossRef]
- Lee, A.S.; Zee, B.C.; Cheung, F.Y.; Kwong, P.; Cheng, A.C.K.; Lai, M.; Kwok, C.; Chong, M.; Jolivet, J.; Chiang, C.L.; et al. Randomized phase II study of the x-linked inhibitor of apoptosis (XIAP) antisense AEG35156 in combination with sorafenib in patients with advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2012, 30, 4105. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, S.; Zhang, Y.; Kmieciak, M.; Leng, Y.; Li, L.; Lin, H.; Turner, J.; Sullivan, D.; Dai, Y.; et al. Abstract 3020: Targeting both canonical and non-canonical NF-kB pathways by the IAP antagonist birinapant potentiates bortezomib anti-myeloma activity. Cancer Res. 2016, 76, 3020. [Google Scholar] [CrossRef]
- Graham, M.A.; Mitsuuchi, Y.; Burns, J.; Chunduru, S.; Benetatos, C.; McKinlay, M.; Weng, D.; Wick, M.J.; Tolcher, A.W.; Papadopoulos, K.; et al. Abstract A25: Phase 1 PK/PD analysis of the Smac-mimetic TL32711 demonstrates potent and sustained cIAP1 suppression in patient PBMCs and tumor biopsies. Apoptosis Necrosis Autophagy 2011, 10, A25. [Google Scholar] [CrossRef]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a Novel Small-Molecule Survivin Suppressant, Induces Regression of Established Human Hormone-Refractory Prostate Tumor Xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.J.; Thomas, A.; Rajan, A.; Chun, G.; Lopez-Chavez, A.; Szabo, E.; Spencer, S.; Carter, C.A.; Guha, U.; Khozin, S.; et al. A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [Google Scholar] [CrossRef]
- Brenke, J.K.; Popowicz, G.M.; Schorpp, K.; Rothenaigner, I.; Roesner, M.; Meininger, I.; Kalinski, C.; Ringelstetter, L.; R’Kyek, O.; Jürjens, G.; et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J. Biol. Chem. 2018, 293, 13191–13203. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Coon, T.; Glasser, J.; McVerry, B.; Zhao, J.; Zhao, Y.; Zou, C.; Ellis, B.; Sciurba, H.; Zhang, Y.; et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 2013, 14, 470–479. [Google Scholar] [CrossRef]
- Mallampalli, R.K.; Coon, T.A.; Glasser, J.R.; Wang, C.; Dunn, S.R.; Weathington, N.M.; Zhao, J.; Zou, C.; Zhao, Y.; Chen, B.B. Targeting F Box Protein Fbxo3 to Control Cytokine-Driven Inflammation. J. Immunol. 2013, 191, 5247–5255. [Google Scholar] [CrossRef] [Green Version]
- Tian, M.; Zeng, T.; Liu, M.; Han, S.; Lin, H.; Lin, Q.; Li, L.; Jiang, T.; Li, G.; Lin, H.; et al. A cell-based high-throughput screening method based on a ubiquitin-reference technique for identifying modulators of E3 ligases. J. Biol. Chem. 2019, 294, 2880–5770. [Google Scholar] [CrossRef] [Green Version]
- Peter, S.; Bultinck, J.; Myant, K.; A Jaenicke, L.; Walz, S.; Müller, J.; Gmachl, M.; Treu, M.; Boehmelt, G.; Ade, C.P.; et al. Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE 1 ubiquitin ligase. EMBO Mol. Med. 2014, 6, 1525–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, L.J.; Campbell, D.C.; Morgan, J.J.; Lawson, M.A.; Down, J.M.; Chauhan, D.; McAvera, R.M.; Morris, T.C.; Hamilton, C.; Krishnan, A.; et al. The E3 ligase HUWE1 inhibition as a therapeutic strategy to target MYC in multiple myeloma. Oncogene 2020, 39, 5001–5014. [Google Scholar] [CrossRef] [PubMed]
- Celegato, M.; Messa, L.; Goracci, L.; Mercorelli, B.; Bertagnin, C.; Spyrakis, F.; Suarez, I.; Cousido-Siah, A.; Travé, G.; Banks, L.; et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. 2020, 470, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.J.; Rietz, A.; Malinkevich, A.; Liu, Y.; Xie, M.; Bartolowits, M.; Davisson, V.J.; Baleja, J.D.; Androphy, E.J. Structure Based Identification and Characterization of Flavonoids That Disrupt Human Papillomavirus-16 E6 Function. PLoS ONE 2013, 8, e84506. [Google Scholar] [CrossRef]
- Strickson, S.; Campbell, D.G.; Emmerich, C.H.; Knebel, A.; Plater, L.; Ritorto, M.S.; Shpiro, N.; Cohen, P. The anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem. J. 2013, 451, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ruan, Y.; Wang, X.; Min, L.; Shen, Z.; Sun, Y.; Qin, X. BAY 11-7082, a nuclear factor-κB inhibitor, induces apoptosis and S phase arrest in gastric cancer cells. J. Gastroenterol. 2013, 49, 864–874. [Google Scholar] [CrossRef]
- Katsuya, K.; Oikawa, D.; Iio, K.; Obika, S.; Hori, Y.; Urashima, T.; Ayukawa, K.; Tokunaga, F. Small-molecule inhibitors of linear ubiquitin chain assembly complex (LUBAC), HOIPINs, suppress NF-κB signaling. Biochem. Biophys. Res. Commun. 2019, 509, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Sato, Y.; Ohtake, F.; Komakura, K.; Hanada, K.; Sugawara, K.; Terawaki, S.; Mizukami, Y.; Phuong, H.T.; Iio, K.; et al. Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses. Commun. Biol. 2020, 3, 163. [Google Scholar] [CrossRef]
- Keating, M.J.; Bach, C.; Yasothan, U.; Kirkpatrick, P. Bendamustine. Nat. Rev. Drug Discov. 2008, 7, 473–474. [Google Scholar] [CrossRef]
- Damaj, G.; Gressin, R.; Bouabdallah, K.; Cartron, G.; Choufi, B.; Gyan, E.; Banos, A.; Jaccard, A.; Park, S.; Tournilhac, O.; et al. Results from a Prospective, Open-Label, Phase II Trial of Bendamustine in Refractory or Relapsed T-Cell Lymphomas: The BENTLY Trial. J. Clin. Oncol. 2013, 31, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, E.; Kim, S.-Z.; Rost, A.; Schuppert, H.; Seipelt, G.; Hoelzer, D.; Mitrou, P.S. Bendamustine is effective in relapsed or refractory aggressive non-Hodgkin’s lymphoma. Ann. Oncol. 2002, 13, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, E.; Neumann, A.; Fauth, F.; Atmaca, A.; Al-Batran, S.E.; Pauligk, C.; Jäger, E. Phase II study of bendamustine in combination with rituximab as first-line treatment in patients 80 years or older with aggressive B-cell lymphomas. Ann. Oncol. 2011, 22, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Panayiotidis, P.; Afanasyev, B.; Janssens, A.; Grosicki, S.; Homenda, W.; Smolej, L.; Kuliczkowski, K.; Doubek, M.; Domnikova, N.; et al. A phase 2, multicenter study investigating ofatumumab and bendamustine combination in patients with untreated or relapsed CLL. Am. J. Hematol. 2016, 91, 900–906. [Google Scholar] [CrossRef]
- Midgley, C.A.; Lane, D.P. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 1997, 15, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Mayat, R.; Kazazt, A.; Orent, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene 2007, 27, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Wang, H.; Li, M.; Rayburn, E.R.; Agrawal, S.; Zhang, R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene 2005, 24, 7238–7247. [Google Scholar] [CrossRef] [Green Version]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53–MDM2 interaction by structure-based design starting from a conformational argument. Bioorg. Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagan, J.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, P.; Inuzuka, H.; Wei, W. Roles of F-box proteins in cancer. Nat. Rev. Cancer 2014, 14, 233–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumimoto, K.; Yamauchi, Y.; Nakayama, K.I. F-Box Proteins and Cancer. Cancers 2020, 12, 1249. [Google Scholar] [CrossRef] [PubMed]
- Aghajan, M.; Jonai, N.; Flick, K.; Fu, F.; Luo, M.; Cai, X.; Ouni, I.; Pierce, N.; Tang, X.; Lomenick, B.; et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 2010, 28, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.-A.; Houghton, P.J. The tor pathway: A target for cancer therapy. Nat. Rev. Cancer 2004, 4, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor Activity of Rapamycin in a Phase I Trial for Patients with Recurrent PTEN-Deficient Glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef]
- Su, J.; Wang, L.; Yin, X.; Zhao, Z.; Hou, Y.; Ye, X.; Zhou, X.; Wang, Z. Rottlerin exhibits anti-cancer effect through inactivation of S phase kinase-associated protein 2 in pancreatic cancer cells. Am. J. Cancer Res. 2016, 6, 2178–2191. [Google Scholar] [PubMed]
- Yin, X.; Zhang, Y.; Su, J.; Hou, Y.; Wang, L.; Ye, X.; Zhao, Z.; Zhou, X.; Li, Y.; Wang, Z. Rottlerin exerts its anti-tumor activity through inhibition of Skp2 in breast cancer cells. Oncotarget 2016, 7, 66512–66524. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yokoyama, N.N.; Zhang, S.; Ding, L.; Liu, H.-M.; Lilly, M.B.; Mercola, D.; Zi, X. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget 2015, 6, 41809–41824. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yu, X.; Li, M.; Gong, G.; Liu, W.; Li, T.; Zuo, H.; Li, W.; Gao, F.; Liu, H. Cdh1-mediated Skp2 degradation by dioscin reprogrammes aerobic glycolysis and inhibits colorectal cancer cells growth. EBioMedicine 2020, 51, 102570. [Google Scholar] [CrossRef] [PubMed]
- Benary, U.; Wolf, J. Wolf Controlling Nuclear NF-κB Dynamics by β-TrCP—Insights from a Computational Model. Biomedicine 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Nalepa, G.; Rolfe, M.; Harper, J. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef]
- Müerköster, S.; Arlt, A.; Sipos, B.; Witt, M.; Großmann, M.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; Schäfer, H. Increased Expression of the E3-Ubiquitin Ligase Receptor Subunit βTRCP1 Relates to Constitutive Nuclear Factor-κB Activation and Chemoresistance in Pancreatic Carcinoma Cells. Cancer Res. 2005, 65, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, H.; Fujiwara, H.; Furuichi, Y.; Tanaka, K.; Shimbara, N. A novel small-molecule inhibitor of NF-κB signaling. Biochem. Biophys. Res. Commun. 2008, 368, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Plechanovová, A.; Jaffray, E.G.; McMahon, S.A.; Johnson, K.A.; Navrátilová, I.; Naismith, J.; Hay, R.T. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011, 18, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; Lacasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef]
- Wu, H.; Tschopp, J.; Lin, S.-C. Smac Mimetics and TNFα: A Dangerous Liaison? Cell 2007, 131, 655–658. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Tcherpakov, M. Will the Ubiquitin System Furnish as Many Drug Targets as Protein Kinases? Cell 2010, 143, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, J.-I.; Ishidab, T.; Tsukamotoa, N.; Kobayashia, N.; Naitoa, A.; Azumaa, S.; Yamamotoa, T. Tumor Necrosis Factor Receptor-Associated Factor (TRAF) Family: Adapter Proteins That Mediate Cytokine Signaling. Exp. Cell Res. 2000, 254, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Henkler, F.; Scheurich, P. The TNF-receptor-associated factor family: Scaffold molecules for cytokine receptors, kinases and their regulators. Cell. Signal. 2001, 13, 389–400. [Google Scholar] [CrossRef]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Zanoni, I.; Granucci, F. Deciphering the complexity of Toll-like receptor signaling. Cell. Mol. Life Sci. 2010, 67, 4109–4134. [Google Scholar] [CrossRef] [PubMed]
- Starczynowski, D.T.; Lockwood, W.W.; Deléhouzée, S.; Chari, R.; Wegrzyn, J.; Fuller, M.; Tsao, M.-S.; Lam, S.; Gazdar, A.F.; Lam, W.L.; et al. TRAF6 is an amplified oncogene bridging the RAS and NF-κB pathways in human lung cancer. J. Clin. Investig. 2011, 121, 4095–4105. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Zheng, M.; Liu, H.; Song, C.; Zhang, W.; Yan, J.; Qin, L.; Liu, X. TRAF6 regulates proliferation, apoptosis, and invasion of osteosarcoma cell. Mol. Cell. Biochem. 2012, 371, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Cao, F.; You, Q. Effect of TRAF6 on the biological behavior of human lung adenocarcinoma cell. Tumor Biol. 2012, 34, 231–239. [Google Scholar] [CrossRef]
- Owais, A.; Mishra, R.K.; Kiyokawa, H. The HECT E3 ligase E6AP/UBE3A as a Therapeutic Target in Cancer and Neurological Disorders. Cancers 2020, 12, 2108. [Google Scholar] [CrossRef]
- Watt, J.E.; Hughes, G.R.; Walpole, S.; Monaco, S.; Stephenson, G.R.; Page, P.C.B.; Hemmings, A.M.; Angulo, J.; Chantry, A. Discovery of Small Molecule WWP2 Ubiquitin Ligase Inhibitors. Chem. A Eur. J. 2018, 24, 17677–17680. [Google Scholar] [CrossRef]
- Wang, M.; Guo, L.; Wu, Q.; Zeng, T.; Lin, Q.; Qiao, Y.; Wang, Q.; Liu, M.; Zhang, X.; Ren, L.; et al. ATR/Chk1/Smurf1 pathway determines cell fate after DNA damage by controlling RhoB abundance. Nat. Commun. 2014, 5, 4901. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.-H.; Wu, H.-T.; Wu, K.-J. Ubiquitination by HUWE1 in tumorigenesis and beyond. J. Biomed. Sci. 2018, 25, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ricci-López, J.; Vidal-Limon, A.; Zunñiga, M.; Jimènez, V.A.; Alderete, J.B.; Brizuela, C.A.; Aguila, S. Molecular modeling simulation studies reveal new potential inhibitors against HPV E6 protein. PLoS ONE 2019, 14, e0213028. [Google Scholar] [CrossRef]
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert Opin. Ther. Patents 2018, 28, 919–937. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural Product-Like Macrocyclic N-Methyl-Peptide Inhibitors against a Ubiquitin Ligase Uncovered from a Ribosome-Expressed De Novo Library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef] [Green Version]
- Mund, T.; Lewis, M.J.; Maslen, S.; Pelham, H.R. Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA 2014, 111, 16736–16741. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Dai, X.; Jiang, W.; Li, Y.; Wei, W. RBR E3 ubiquitin ligases in tumorigenesis. Semin. Cancer Biol. 2020, 67, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.-L.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nat. Cell Biol. 2011, 471, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; Van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nat. Cell Biol. 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.-I.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nat. Cell Biol. 2011, 471, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Schmitz, R.; Mitala, J.; Whiting, A.L.; Xiao, W.; Ceribelli, M.; Wright, G.W.; Zhao, H.; Yang, Y.; Xu, W.; et al. Essential Role of the Linear Ubiquitin Chain Assembly Complex in Lymphoma Revealed by Rare Germline Polymorphisms. Cancer Discov. 2014, 4, 480–493. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin Suppresses NF-κB Activation by Selectively Inhibiting Linear Ubiquitin Chain Assembly Complex (LUBAC). ACS Chem. Biol. 2015, 10, 675–681. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, V.; Johnson, C.; Barlow, V.; Hastie, J.; Knebel, A.; Trost, M. The MALDI-TOF E2/E3 Ligase Assay as Universal Tool for Drug Discovery in the Ubiquitin Pathway. Cell Chem. Biol. 2018, 25, 1117–1127.e4. [Google Scholar] [CrossRef] [Green Version]
- Johansson, H.; Tsai, Y.-C.I.; Fantom, K.; Chung, C.-W.; Kümper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.I.; Johansson, H.; Dixon, D.; Martin, S.; Chung, C.-W.; Clarkson, J.; House, D.; Rittinger, K. Single-Domain Antibodies as Crystallization Chaperones to Enable Structure-Based Inhibitor Development for RBR E3 Ubiquitin Ligases. Cell Chem. Biol. 2020, 27, 83–93.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eletr, Z.M.; Wilkinson, K.D. Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sidhu, S.S. Development of inhibitors in the ubiquitination cascade. FEBS Lett. 2014, 588, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nat. Cell Biol. 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A Dynamic Role of HAUSP in the p53-Mdm2 Pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W.; et al. Discovery of Specific Inhibitors of Human USP7/HAUSP Deubiquitinating Enzyme. Chem. Biol. 2012, 19, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstock, J.; Wu, J.; Cao, P.; Kingsbury, W.D.; McDermott, J.L.; Kodrasov, M.P.; McKelvey, D.M.; Kumar, K.G.S.; Goldenberg, S.J.; Mattern, M.R.; et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med. Chem. Lett. 2012, 3, 789–792. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.-H.; Cheng, J.; A Vasudevan, S.; Dou, J.; Zhang, H.; Patel, R.H.; Ma, I.T.; Rojas, Y.; Zhao, Y.; Yu, Y.; et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013, 4, e867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altun, M.; Kramer, H.B.; Willems, L.I.; McDermott, J.L.; Leach, C.A.; Goldenberg, S.J.; Kumar, K.S.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem. Biol. 2011, 18, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Pozhidaeva, A.; Valles, G.; Wang, F.; Wu, J.; Sterner, D.E.; Nguyen, P.; Weinstock, J.; Kumar, K.S.; Kanyo, J.; Wright, D.; et al. USP7-Specific Inhibitors Target and Modify the Enzyme’s Active Site via Distinct Chemical Mechanisms. Cell Chem. Biol. 2017, 24, 1501–1512.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavana, O.; Gu, W. Modulation of the p53/MDM2 interplay by HAUSP inhibitors. J. Mol. Cell Biol. 2017, 9, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavory, G.; O’Dowd, C.R.; Helm, M.D.; Flasz, J.; Arkoudis, E.; Dossang, A.; Hughes, C.; Cassidy, E.; McClelland, K.; Odrzywol, E.; et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 2018, 14, 118–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lello, P.; Pastor, R.; Murray, J.M.; Blake, R.A.; Cohen, F.; Crawford, T.D.; Drobnick, J.; Drummond, J.; Kategaya, L.; Kleinheinz, T.; et al. Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques. J. Med. Chem. 2017, 60, 10056–10070. [Google Scholar] [CrossRef] [PubMed]
- Lamberto, I.; Liu, X.; Seo, H.-S.; Schauer, N.J.; Iacob, R.E.; Hu, W.; Das, D.; Mikhailova, T.; Weisberg, E.L.; Engen, J.R.; et al. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem. Biol. 2017, 24, 1490–1500.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dexheimer, T.S.; Ai, Y.; Liang, Q.; Villamil, M.A.; Inglese, J.; Maloney, D.J.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Selective and Cell-Active Inhibitors of the USP1/ UAF1 Deubiquitinase Complex Reverse Cisplatin Resistance in Non-small Cell Lung Cancer Cells. Chem. Biol. 2011, 18, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, E.; Hwang, D.; Bheda-Malge, A.; Whitehurst, C.B.; Kabanov, A.V.; Kondo, S.; Aga, M.; Yoshizaki, T.; Pagano, J.S.; Sokolsky, M.; et al. Inhibition of UCH-L1 Deubiquitinating Activity with Two Forms of LDN-57444 Has Anti-Invasive Effects in Metastatic Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 3733. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.F.; Sun, H.; Liu, Y.; Potu, H.; Kandarpa, M.; Ermann, M.; Courtney, S.M.; Young, M.; Showalter, H.D.; Sun, D.; et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015, 125, 3588–3597. [Google Scholar] [CrossRef]
- Clancy, A.; Heride, C.; Pinto-Fernández, A.; Elcocks, H.; Kallinos, A.; Kayser-Bricker, K.J.; Wang, W.; Smith, V.; Davis, S.; Fessler, S.; et al. The deubiquitylase USP9X controls ribosomal stalling. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Zhang, W.; Sidhu, S.S. Allosteric inhibitors hit USP7 hard. Nat. Chem. Biol. 2018, 14, 110–111. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nat. Cell Biol. 2017, 550, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Cohn, M.A.; Kowal, P.; Yang, K.; Haas, W.; Huang, T.T.; Gygi, S.P.; D’Andrea, A.D. A UAF1-Containing Multisubunit Protein Complex Regulates the Fanconi Anemia Pathway. Mol. Cell 2007, 28, 786–797. [Google Scholar] [CrossRef]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef] [Green Version]
- Boselli, M.; Lee, B.-H.; Robert, J.; Prado, M.A.; Min, S.-W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.L.; De Jong, A.; Leestemaker, Y.; Geurink, P.P.; Wijdeven, R.H.; Ovaa, H.; Dolan, B.P. Inhibition of the Deubiquitinase Usp14 Diminishes Direct MHC Class I Antigen Presentation. J. Immunol. 2018, 200, 928–936. [Google Scholar] [CrossRef]
- Sha, B.; Chen, X.; Wu, H.; Li, M.; Shi, J.; Wang, L.; Liu, X.; Chen, P.; Hu, T.; Li, P. Deubiquitylatinase inhibitor b-AP15 induces c-Myc-Noxa-mediated apoptosis in esophageal squamous cell carcinoma. Apoptosis 2019, 24, 826–836. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., III; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 Metalloprotease in Deubiquitination and Degradation by the 26S Proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Perez, C.; Li, J.; Parlati, F.; Rouffet, M.; Ma, Y.; MacKinnon, A.L.; Chou, T.-F.; Deshaies, R.J.; Cohen, S.M.; Parlati, F.; et al. Discovery of an Inhibitor of the Proteasome Subunit Rpn11. J. Med. Chem. 2017, 60, 1343–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauinger, L.; Li, J.; Shostak, A.; Cemel, I.A.; Ha, N.; Zhang, Y.; Merkl, P.E.; Obermeyer, S.; Stankovic-Valentin, N.; Schafmeier, T.; et al. Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat. Chem. Biol. 2017, 13, 709–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, C.A.; Baljinnyam, B.; Zakharov, A.V.; Jadhav, A.; Simeonov, A.; Zhuang, Z. Cell Lysate-Based AlphaLISA Deubiquitinase Assay Platform for Identification of Small Molecule Inhibitors. ACS Chem. Biol. 2017, 12, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lashuel, H.A.; Choi, S.; Xing, X.; Case, A.; Ni, J.; Yeh, L.-A.; Cuny, G.D.; Stein, R.L.; Lansbury, P.T. Discovery of Inhibitors that Elucidate the Role of UCH-L1 Activity in the H1299 Lung Cancer Cell Line. Chem. Biol. 2003, 10, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Kooij, R.; Liu, S.; Sapmaz, A.; Xin, B.-T.; Janssen, G.M.C.; Van Veelen, P.A.; Ovaa, H.; Dijke, P.T.; Geurink, P. A Small-Molecule Activity-Based Probe for Monitoring Ubiquitin C-terminal Hydrolase L1 (UCHL1) Activity in Live Cells and Zebrafish Embryos. J. Am. Chem. Soc. 2020, 142, 16825–16841. [Google Scholar] [CrossRef]
- Peddaboina, C.; Jupiter, D.; Fletcher, S.; Yap, J.L.; Rai, A.; Tobin, R.P.; Jiang, W.; Rascoe, P.; Rogers, M.K.N.; Smythe, W.R.; et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer 2012, 12, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nat. Cell Biol. 2009, 463, 103–107. [Google Scholar] [CrossRef]
- Kapuria, V.; Levitzki, A.; Bornmann, W.G.; Maxwell, D.; Priebe, W.; Sorenson, R.J.; Showalter, H.D.; Talpaz, M.; Donato, N.J. A novel small molecule deubiquitinase inhibitor blocks Jak2 signaling through Jak2 ubiquitination. Cell. Signal. 2011, 23, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kollipara, R.K.; Srivastava, N.; Li, R.; Ravindranathan, P.; Hernandez, E.; Freeman, E.; Humphries, C.G.; Kapur, P.; Lotan, Y.; et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4251–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zheng, H.; Mao, A.-P.; Zhong, B.; Li, Y.; Liu, Y.; Gao, Y.; Ran, Y.; Tien, P.; Shu, H.-B. Regulation of Virus-triggered Signaling by OTUB1- and OTUB2-mediated Deubiquitination of TRAF3 and TRAF6. J. Biol. Chem. 2010, 285, 4291–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Vinik, Y.; Shatz-Azoulay, H.; Isaac, R.; Streim, S.; Jona, G.; Boura-Halfon, S.; Zick, Y. Otubain 2 is a novel promoter of beta cell survival as revealed by siRNA high-throughput screens of human pancreatic islets. Diabetologia 2013, 56, 1317–1326. [Google Scholar] [CrossRef]
- Kudo, L.C.; Parfenova, L.; Vi, N.; Lau, K.; Pomakian, J.; Valdmanis, P.; Rouleau, G.A.; Vinters, H.V.; Wiedau-Pazos, M.; Karsten, S.L. Integrative gene–tissue microarray-based approach for identification of human disease biomarkers: Application to amyotrophic lateral sclerosis. Hum. Mol. Genet. 2010, 19, 3233–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Du, J.; Wang, S.; Shao, L.; Jin, K.; Li, F.; Wei, B.; Ding, W.; Fu, P.; van Dam, H.; et al. OTUB2 Promotes Cancer Metastasis via Hippo-Independent Activation of YAP and TAZ. Mol. Cell 2019, 73, 7–21.e7. [Google Scholar] [CrossRef] [Green Version]
- Mevissen, T.E.; Hospenthal, M.K.; Geurink, P.; Elliott, P.; Akutsu, M.; Arnaudo, N.; Ekkebus, R.; Kulathu, Y.; Wauer, T.; El Oualid, F.; et al. OTU Deubiquitinases Reveal Mechanisms of Linkage Specificity and Enable Ubiquitin Chain Restriction Analysis. Cell 2013, 154, 169–184. [Google Scholar] [CrossRef] [Green Version]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [Green Version]
- Bednash, J.S.; Weathington, N.; Londino, J.; Rojas, M.; Gulick, D.L.; Fort, R.; Han, S.; McKelvey, A.C.; Chen, B.B.; Mallampalli, R.K. Targeting the deubiquitinase STAMBP inhibits NALP7 inflammasome activity. Nat. Commun. 2017, 8, 15203. [Google Scholar] [CrossRef] [Green Version]
- Aleo, E.; Henderson, C.J.; Fontanini, A.; Solazzo, B.; Brancolini, C. Identification of New Compounds That Trigger Apoptosome-Independent Caspase Activation and Apoptosis. Cancer Res. 2006, 66, 9235–9244. [Google Scholar] [CrossRef] [Green Version]
- Reiner, T.; Parrondo, R.; Pozas, A.D.L.; Palenzuela, D.; Perez-Stable, C. Betulinic Acid Selectively Increases Protein Degradation and Enhances Prostate Cancer-Specific Apoptosis: Possible Role for Inhibition of Deubiquitinase Activity. PLoS ONE 2013, 8, e56234. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. Identifying druggable disease-modifying gene products. Curr. Opin. Chem. Biol. 2009, 13, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Russ, A.P.; Lampel, S. The druggable genome: An update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Ottis, P.; Crews, C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017, 12, 892–898. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. Targeted Protein Degradation: From Chemical Biology to Drug Discovery. Cell Chem. Biol. 2017, 24, 1181–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. 2016, 55, 1966–1973. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nat. Cell Biol. 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, present and future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Bondeson, D.P.; Mares, A.; Stafford-Smith, M.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef] [PubMed]
- Leiser, D.; Pochon, B.; Blank-Liss, W.; Francica, P.; Glück, A.A.; Aebersold, D.M.; Zimmer, Y.; Medová, M. Targeting of the MET receptor tyrosine kinase by small molecule inhibitors leads to MET accumulation by impairing the receptor downregulation. FEBS Lett. 2014, 588, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.; Cromm, P.; Zimmermann, G.; Grossmann, T.; Waldmann, H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. [Google Scholar] [CrossRef]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the IkBa-ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, J.S.; Fonseca, F.N.; Koldobskiy, M.; Mandal, A.; Deshaies, R.; Sakamoto, K.M.; Crews, C.M. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J. Am. Chem. Soc. 2004, 126, 3748–3754. [Google Scholar] [CrossRef] [Green Version]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules to Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Molle, I.; Thomann, A.; Buckley, D.L.; So, E.C.; Lang, S.; Crews, C.M.; Ciulli, A. Dissecting Fragment-Based Lead Discovery at the von Hippel-Lindau Protein:Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem. Biol. 2012, 19, 1300–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdeano, C.; Gadd, M.S.; Soares, P.; Scaffidi, S.; Van Molle, I.; Birced, I.; Hewitt, S.; Dias, D.M.; Ciulli, A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem. 2014, 57, 8657–8663. [Google Scholar] [CrossRef]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkina, A.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nat. Cell Biol. 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Baratta, M.G.; Schinzel, A.C.; Zwang, Y.; Bandopadhayay, P.; Bowman-Colin, C.; Kutt, J.; Curtis, J.; Piao, H.; Wong, L.C.; Kung, A.L.; et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.; Buckley, D.L.; Paulk, J.; Roberts, J.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Ishikawa, M.; Tomoshige, S.; Demizu, Y.; Naito, M. Selective Degradation of Target Proteins by Chimeric Small-Molecular Drugs, PROTACs and SNIPERs. Pharmaceuticals 2020, 13, 74. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, A.; Pandiella, A. Proteolysis targeting chimeras (PROTACs) in cancer therapy. J. Exp. Clin. Cancer Res. 2020, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Robb, C.M.; Contreras, J.I.; Kour, S.; Taylor, M.A.; Abid, M.; Sonawane, Y.A.; Zahid, M.; Murry, D.J.; Natarajan, A.; Rana, S. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 2017, 53, 7577–7580. [Google Scholar] [CrossRef]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaime-Figueroa, S.; Buhimschi, A.D.; Toure, M.; Hines, J.; Crews, C.M. Design, synthesis and biological evaluation of Proteolysis Targeting Chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2020, 30, 126877. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorg. Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef]
- Bassi, Z.I.; Fillmore, M.C.; Miah, A.H.; Chapman, T.D.; Maller, C.; Roberts, E.J.; Davis, L.C.; Lewis, D.E.; Galwey, N.W.; Waddington, K.E.; et al. Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem. Biol. 2018, 13, 2862–2867. [Google Scholar] [CrossRef] [PubMed]
- McCoull, W.; Cheung, T.; Anderson, E.; Barton, P.; Burgess, J.; Byth, K.; Cao, Q.; Castaldi, M.P.; Chen, H.; Chiarparin, E.; et al. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC to Provide Insight into Small Molecule Targeting of BCL. ACS Chem. Biol. 2018, 13, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Bai, L.; Xu, R.; Zhao, Y.; Chen, J.; McEachern, D.; Chinnaswamy, K.; Wen, B.; Dai, L.; Kumar, P.; et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J. Med. Chem. 2019, 62, 11280–11300. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, B.; Yang, C.-Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. 2016, 55, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.-H.; Chen, W.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation Accession codes Atomic coordinates and structure factors for hsBrd4 BD2-MZ1-hsVHL-hsEloC-hsEloB have been deposited in the Protein Data Bank (PDB) under accession number. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Crew, A.P.; Raina, K.; Dong, H.; Qian, Y.; Wang, J.; Vigil, D.; Serebrenik, Y.; Hamman, B.D.; Morgan, A.; Ferraro, C.; et al. Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase. J. Med. Chem. 2018, 61, 583–598. [Google Scholar] [CrossRef]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.A.; Reyes, J.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.G.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Kang, C.H.; Lee, D.H.; Lee, C.O.; Du Ha, J.; Park, C.H.; Hwang, J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 2018, 505, 542–547. [Google Scholar] [CrossRef]
- Cromm, P.M.; Samarasinghe, K.T.G.; Hines, J.; Crews, C.M. Addressing Kinase-Independent Functions of Fak via PROTAC-Mediated Degradation. J. Am. Chem. Soc. 2018, 140, 17019–17026. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Qin, C.; Xiang, W.; Fernandez-Salas, E.; Yang, C.-Y.; Wang, M.; Zhao, L.; Xu, T.; Chinnaswamy, K.; et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J. Med. Chem. 2019, 62, 941–964. [Google Scholar] [CrossRef]
- Smith, B.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, J.; McGonagle, G.A.; Eden, J.; Kiritharan, G.; Touzet, M.; Lewell, X.; Emery, J.; Eidam, H.; Harling, J.D.; Anderson, N.A. Targeting IRAK4 for Degradation with PROTACs. ACS Med. Chem. Lett. 2019, 10, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Zhang, X.; Thummuri, D.; Liu, X.; Hu, W.; Zhang, P.; Khan, S.; Yuan, Y.; Zhou, D.; Zheng, G. Discovery of PROTAC BCL-XL degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem. 2020, 192, 112186. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of apoptosis in MDA-MB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2019, 62, 448–466. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
- Spradlin, J.N.; Hu, X.; Ward, C.C.; Brittain, S.M.; Jones, M.D.; Ou, L.; To, M.; Proudfoot, A.; Ornelas, E.; Woldegiorgis, M.; et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.; Snyder, L.B.; Willard, R.R.; Vitale, N.; Raina, K.; Pizzano, J.; Gordon, D.; Bookbinder, M.; Macaluso, J.; Dong, H.; et al. Abstract 5236: ARV-110: An androgen receptor PROTAC degrader for prostate cancer. Clin. Trials 2018, 78, 5236. [Google Scholar] [CrossRef]
- Flanagan, J.; Qian, Y.; Gough, S.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Poster Sess. Abstr. 2019, 79, P5-04. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Naito, M.; Ohoka, N.; Shibata, N. SNIPERs—Hijacking IAP activity to induce protein degradation. Drug Discov. Today Technol. 2019, 31, 35–42. [Google Scholar] [CrossRef]
- Itoh, Y.; Ishikawa, M.; Kitaguchi, R.; Sato, S.; Naito, M.; Hashimoto, Y. Development of target protein-selective degradation inducer for protein knockdown. Bioorg. Med. Chem. 2011, 19, 3229–3241. [Google Scholar] [CrossRef]
- Okuhira, K.; Demizu, Y.; Hattori, T.; Ohoka, N.; Shibata, N.; Nishimaki-Mogami, T.; Okuda, H.; Kurihara, M.; Naito, M. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013, 104, 1492–1498. [Google Scholar] [CrossRef]
- Ohoka, N.; Morita, Y.; Nagai, K.; Shimokawa, K.; Ujikawa, O.; Fujimori, I.; Ito, M.; Hayase, Y.; Okuhira, K.; Shibata, N.; et al. Derivatization of inhibitor of apoptosis protein (IAP) ligands yields improved inducers of estrogen receptor α degradation. J. Biol. Chem. 2018, 293, 6776–6790. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Nagai, K.; Morita, Y.; Ujikawa, O.; Ohoka, N.; Hattori, T.; Koyama, R.; Sano, O.; Imaeda, Y.; Nara, H.; et al. Development of Protein Degradation Inducers of Androgen Receptor by Conjugation of Androgen Receptor Ligands and Inhibitor of Apoptosis Protein Ligands. J. Med. Chem. 2018, 61, 543–575. [Google Scholar] [CrossRef]
- Ohoka, N.; Ujikawa, O.; Shimokawa, K.; Sameshima, T.; Shibata, N.; Hattori, T.; Nara, H.; Cho, N.; Naito, M. Different Degradation Mechanisms of Inhibitor of Apoptosis Proteins (IAPs) by the Specific and Nongenetic IAP-Dependent Protein Eraser (SNIPER). Chem. Pharm. Bull. 2019, 67, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinebach, C.; Lindner, S.; Udeshi, N.D.; Mani, D.C.; Kehm, H.; Köpff, S.; Carr, S.A.; Gütschow, M.; Krönke, J. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 2018, 13, 2771–2782. [Google Scholar] [CrossRef]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-Induced Protein Degradation with Photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, P.; Samarasinghe, K.T.G.; Crews, C.M.; Carreira, E.M. Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Cent. Sci. 2019, 5, 1682–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, 5064. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H. Ümit; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [Green Version]
- Moreau, K.; Coen, M.; Zhang, A.X.; Pachl, F.; Castaldi, M.P.; Dahl, G.; Boyd, H.; Scott, C.; Newham, P. Proteolysis-targeting chimeras in drug development: A safety perspective. Br. J. Pharmacol. 2020, 177, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: Recent progress and future challenges. Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.F.; Scott-Stevens, P.; Gaohua, L. Targeted protein degradation in vivo with Proteolysis Targeting Chimeras: Current status and future considerations. Drug Discov. Today Technol. 2019, 31, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Roy, R.D.; Rosenmund, C.; Stefan, M. Cooperative binding mitigates the high-dose hook effect. BMC Syst. Biol. 2017, 11, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Troup, R.I.; Fallan, C.; Baud, M.G.J. Current strategies for the design of PROTAC linkers: A critical review. Explor. Target. Anti-tumor Ther. 2020, 1, 273–312. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; Macbeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Lentzsch, S. The application and biology of immunomodulatory drugs (IMiDs) in cancer. Pharmacol. Ther. 2012, 136, 56–68. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nat. Cell Biol. 2014, 512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.F.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2018, 25, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164, 384 Reduces Cellular Estrogen Receptor Content by Increasing Its Turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Lim, S.M.; Westover, K.D.; Dodge, M.E.; Ercan, D.; Ficarro, S.B.; Udayakumar, D.; Gurbani, D.; Tae, H.S.; Riddle, S.M.; et al. Pharmacological targeting of the pseudokinase Her3. Nat. Chem. Biol. 2014, 10, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, J.L.; Neklesa, T.K.; Cox, C.S.; Roth, A.G.; Buckley, D.L.; Tae, H.S.; Sundberg, T.B.; Stagg, D.; Hines, J.; McDonnell, D.P.; et al. Small-Molecule-Mediated Degradation of the Androgen Receptor through Hydrophobic Tagging. Angew. Chem. Int. Ed. 2015, 54, 9659–9662. [Google Scholar] [CrossRef]
- Portnoff, A.D.; Stephens, E.A.; Varner, J.D.; DeLisa, M.P. Ubiquibodies, Synthetic E3 Ubiquitin Ligases Endowed with Unnatural Substrate Specificity for Targeted Protein Silencing. J. Biol. Chem. 2014, 289, 7844–7855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.J.C.; Gollapalli, D.R.; Hedstrom, L. Inhibitor Mediated Protein Degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Kanner, S.A.; Shuja, Z.; Choudhury, P.; Jain, A.; Colecraft, H.M. Targeted deubiquitination rescues distinct trafficking-deficient ion channelopathies. Nat. Methods 2020, 17, 1245–1253. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Avvakumov, G.; Tong, J.; Fan, Y.; Zhao, Y.; Alberts, P.; Persaud, A.; Walker, J.R.; Neculai, A.-M.; Neculai, D.; et al. A Strategy for Modulation of Enzymes in the Ubiquitin System. Science 2013, 339, 590–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, A. Engineering ubiquitin to modulate the ubiquitin proteosome system. Cell Cycle 2013, 12, 1651–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Rouge, L.; Phillips, A.H.; Lam, C.; Liu, P.; Sandoval, W.; Helgason, E.; Murray, J.M.; E Wertz, I.; et al. Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat. Chem. Biol. 2012, 9, 51–58. [Google Scholar] [CrossRef]
- Zhang, W.; Sartori, M.A.; Makhnevych, T.; Federowicz, K.E.; Dong, X.; Liu, L.; Nim, S.; Dong, A.; Yang, J.; Li, Y.; et al. Generation and Validation of Intracellular Ubiquitin Variant Inhibitors for USP7 and USP10. J. Mol. Biol. 2017, 429, 3546–3560. [Google Scholar] [CrossRef]
- Garg, P.; Ceccarelli, D.F.; Keszei, A.F.; Kurinov, I.; Sicheri, F.; Sidhu, S.S. Structural and Functional Analysis of Ubiquitin-based Inhibitors That Target the Backsides of E2 Enzymes. J. Mol. Biol. 2020, 432, 952–966. [Google Scholar] [CrossRef]
- LeBlanc, N.; Mallette, E.; Zhang, W. Targeted modulation of E3 ligases using engineered ubiquitin variants. FEBS J. 2021, 288, 2143–2165. [Google Scholar] [CrossRef] [PubMed]


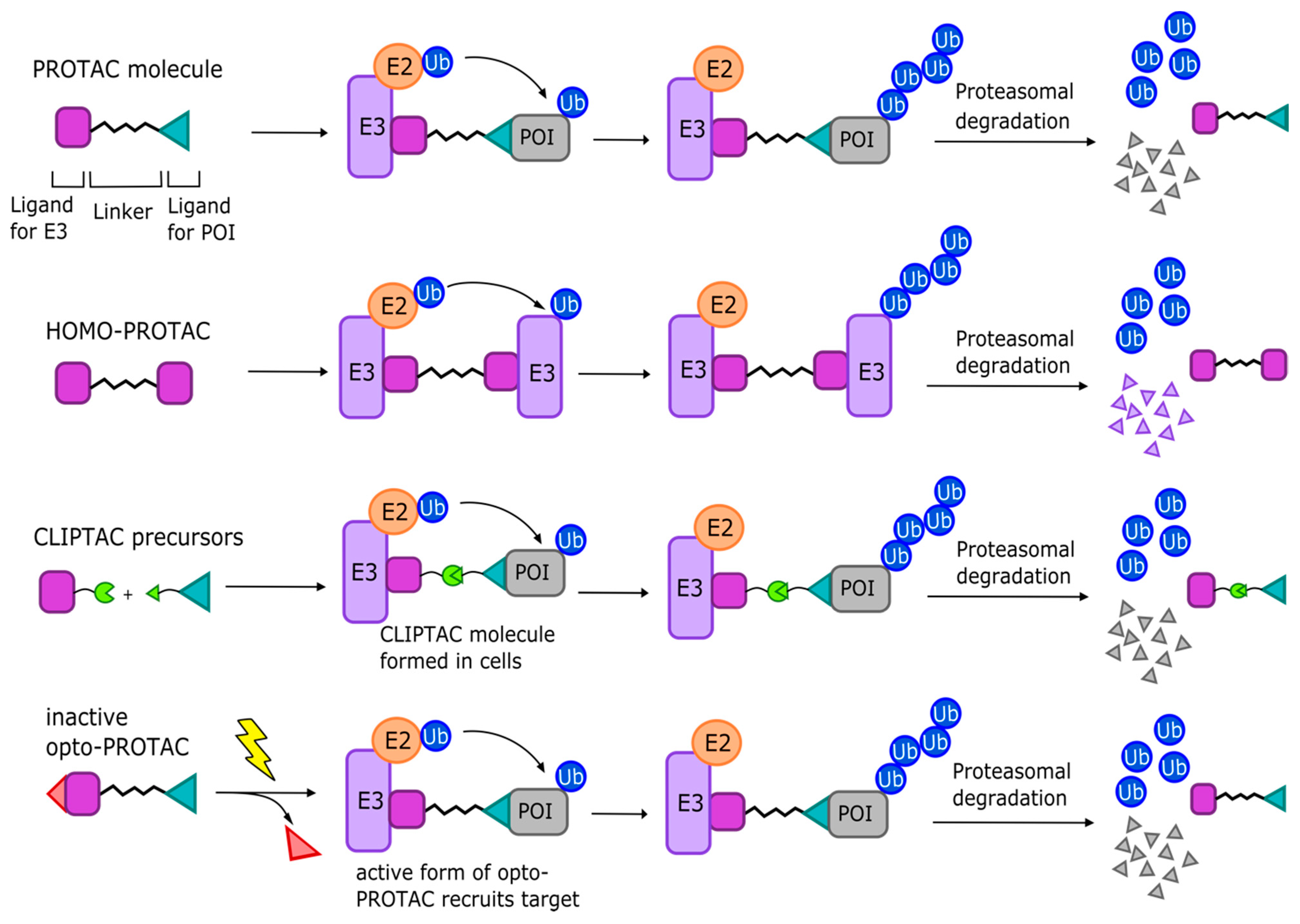

{kind=link}
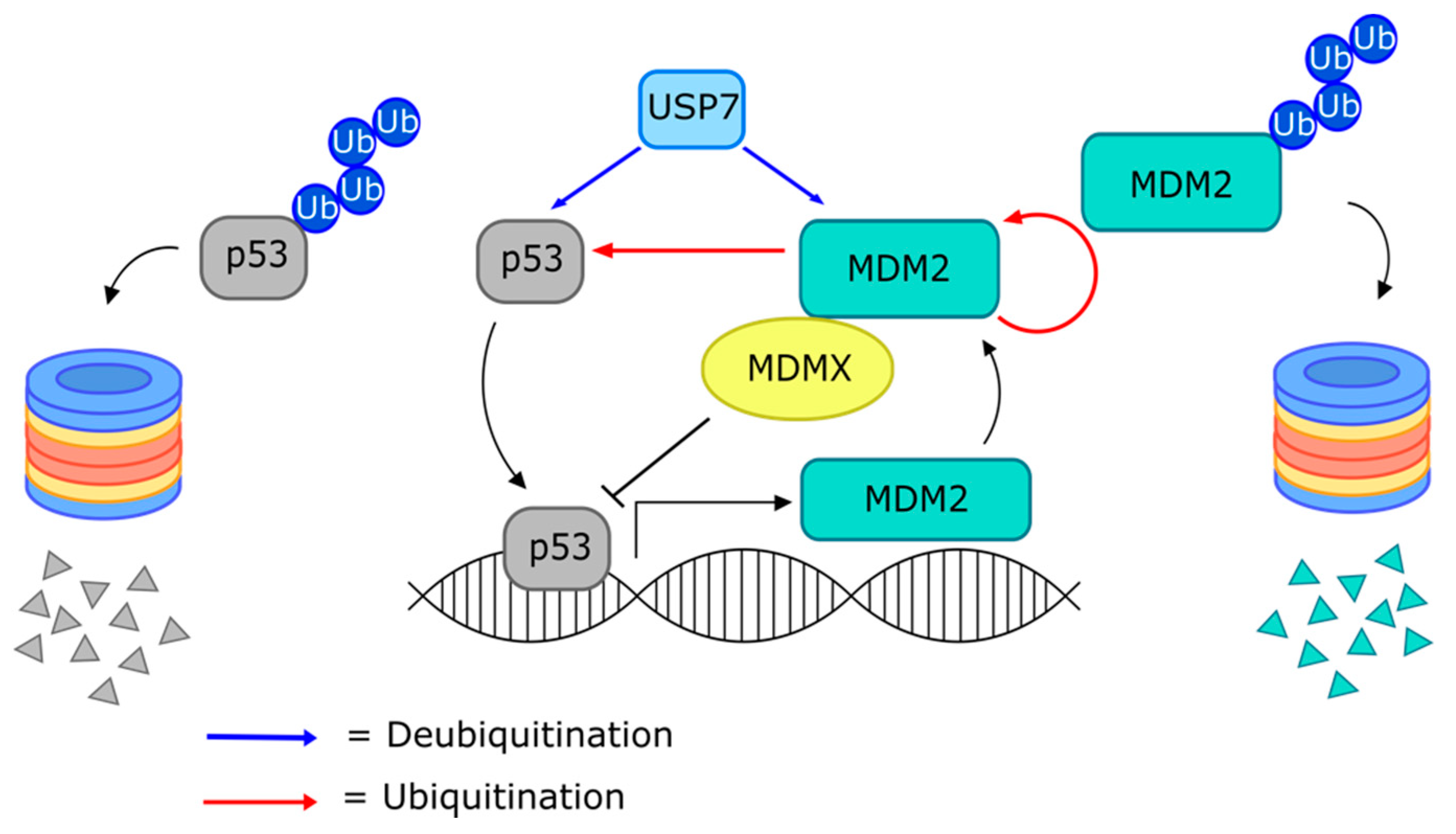{kind=link}
{kind=link}
| Compound Name | UPS Target | Mode of Action | Current Clinical Stage | Cancer Models Targeted in Preclinical Studies | Targeted Cancer Types in Clinical Trials | Clinical Trial Results |
|---|---|---|---|---|---|---|
| Proteasome Inhibitors | ||||||
| Bortezomib | Proteasome (20S particle) | Reversibly binds active sites of 20S proteasome | FDA approved (Phase 4) | MM, lymphocytic leukemia cells, oral squamous carcinoma cells, and more [32] | MM, AML, colorectal cancer, head and neck cancer, mantle cell lymphoma, and more | [59,60,61] |
| Carfilzomib | Proteasome (20S particle) | Irreversibly binds active sites of 20S proteasome | FDA approved (Phase 4) | Hematologic tumors [37] | MM, lymphoma, thyroid cancer, lung cancer, kidney cancer, and more | [36,62,63,64] |
| Oprozomib | Proteasome (20S particle) | Irreversibly binds active sites of 20S proteasome | Phase 1b/2 | MM [65] | Solid tumors, hepatocellular carcinoma, MM, waldenstrom macroglobulinemia, and more | [66] |
| Ixazomib | Proteasome (20S particle) | Inhibits chymotrypsin-like activity of 20S proteasome | FDA approved (Phase 4) | Solid and hematologic tumors [67], MM [68], B-cell, plasma cell malignancies [69] | MM, relapsed/refractory MM, AML, Hodgkin and T-cell lymphoma, breast cancer, and more | [70,71] |
| IU1, IU1-47, 1B10, IU1-248 | Proteasome (19S particle) | Inhibit USP14 (proteasome-associated DUB) via steric site | Preclinical | N/A | N/A | N/A |
| b-AP15 | Proteasome (19S particle) | Targets UCHL5 and USP14 (proteasome-associated DUBs) | Preclinical | Solid tumors, MM [40,72,73,74] | N/A | N/A |
| VLX1570 | Proteasome (19S particle) | Analog of b-AP15, also targets UCHL5 and USP14 | Preclinical (Phase 1/2 terminated) | MM [42] | MM | N/A |
| WP1130 | Proteasome (19S particle) | Inhibits USP9X, UCHL, and USP14 | Preclinical | Chronic myelogenous leukemia (CML), melanoma, glioblastoma, myeproliferative disorders [44,75,76], MCL [77], AML [78] | N/A | N/A |
| RA-9 | Proteasome (19S particle) | Inhibits proteasome-associated DUBs | Preclinical | Ovarian cancer [43] | N/A | N/A |
| RA190 | Proteasome (19S particle) | Inhibits RNP13 and inactivates Uch37 | Preclinical | MM, ovarian cancer [45] | N/A | N/A |
| Capzimin, 8TQ | Proteasome (19S particle) | Specifically target RPN11 | Preclinical | Leukemia, NSCLC, breast cancer [79] | N/A | N/A |
| Thiolutin, SOP11 | Proteasome (19S particle) | Targets RPN11 and other JAMM-family DUBs | Preclinical | Colon cancer, bortezomib-resistant RPE cells [80] | N/A | N/A |
| E1 inhibitors | ||||||
| PYR-41 | UBA1 | Irreversibly binds active site cysteine of UBA1 | Preclinical | N/A | N/A | N/A |
| MLN4942 (Pevonedistat) | NAE | Binds NEDD8, prevents CRL neddylation to inhibit activity | Phase 3 | Colon cancer, lung cancer, myeloma, lymphoma [81,82,83] | AML, MM, lymphoma, melanoma, lung cancer, PCM, and more | [84,85,86,87] |
| E2 inhibitors | ||||||
| CC0651 | hCdc34 | Binds allosteric pocket causing structural displacement | Preclinical | Prostate and colorectal cancer cell lines [88] | N/A | N/A |
| NSC697923 | Ubc-Uev1A | Impedes formation of Ubc13 and Ub thioester conjugate | Preclinical | Diffuse large B-cell lymphoma (DLBCL) [89] | N/A | N/A |
| Compound Name | UPS Target | Mode of Action | Current Clinical Stage | Cancer Models Targeted in Preclinical Studies | Targeted Cancer Types in Clinical Trials | Published Clinical Trial Results |
|---|---|---|---|---|---|---|
| MDM2 inhibitors | ||||||
| Nutlin-3 | MDM2 | Competitive inhibitor of p53 binding site on MDM2 | Preclinical | AML [101], hematologic malignancies [102,103,104,105], breast cancers [106,107], glioblastoma [108] | N/A | N/A |
| RG7112 (RO5045337) | MDM2 | Nutlin derivative, inhibits MDM2–p53 binding site | Phase 1 | Cancer cell lines expressing wild-type p53 [109] | Leukemia, hematologic neoplasms, liposarcoma, advanced solid tumors, myeloproliferative neoplasms | [110,111,112] |
| RG7388 (RO5503781, Idasanutlin) | MDM2 | Nutlin derivative, inhibits MDM2–p53 binding site | Phase 2 (Phase 3 terminated) | Cancer cell lines expressing wild-type p53, osteosarcoma xenografts [113] | AML, ALL, solid tumors, neuroblastoma, plasma cell myeloma, breast cancer, PV and ET, and more | [114,115,116] |
| MI-77301 (SAR405838) | MDM2 | Selectively binds MDM2–p53 binding site, improved affinity via MDM2 N-term refolding | Phase 1 | Leukemia, osteosarcoma, prostate and colon cancer cell lines [117] | Malignant neoplasms, advanced solid tumors, lymphoma | [118,119] |
| MK-8242 (SCH-900242) | MDM2 | Oral MDM2–p53 inhibitor | Phase 1 | N/A | AML, advanced solid tumors | [120,121] |
| AMG 232 | MDM2 | Inhibits MDM2–p53 interaction with improved potency due to hydrophobic interactions with MDM2 “glycine shelf” | Phase 1 | Various tumor cell lines and xenografts [122,123,124] | AML, advanced solid tumors, glioblastoma, gliosarcoma, metastatic melanoma, MM, and more | [125,126] |
| Ds3032b (Milademetan) | MDM2 | Oral MDM2–p53 inhibitor | Phase 2 | Neuroblastoma [127], BCL [10] | Myeloma, AML, recurrent/refractory myeloid leukemia, advanced solid tumors, lymphomas | [128,129] |
| HDM 201 (Siremadlin) | MDM2 | Binds to MDM2, inhibits interaction with p53 | Phase 2 | Wild-type-p53 cancer cell lines [130] | AML, colorectal cancer, liposarcoma, malignant solid tumors, and more | [131,132] |
| APG-115 | MDM2 | Oral MDM2–p53 inhibitor | Phase 2 | Osteosarcoma xenografts [133] | AML, T-prolymphocytic leukemia, liposarcoma, advanced solid tumors, melanoma, salivary gland cancer | [134,135] |
| CGM097 | MDM2 | MDM2–p53 inhibitor | Phase 1 | Colorectal cancer, osteosarcoma cells [136] | Solid tumors with wild-type p53 | N/A |
| PRIMA1, APR-246 | MDM2 | Binds core domain of p53, preventing MDM2 association | Phase 3 | Osteosarcoma, NSCLC, adenocarcinoma cell lines and xenografts [137,138], myeloma [139] | AML, myeloid malignancies, NSCLC, gastric cancer, esophageal carcinoma, non-Hodgkin’s lymphoma, CLL, MCL, hematologic neoplasms, and more | [140] |
| MI-63 | MDM2 | Binds MDM2, preventing p53 association | Preclinical | Prostate cancer cells with wt-p53 [141] | N/A | N/A |
| MI-219 | MDM2 | Binds MDM2, preventing p53 association; can also induce degradation of MDMX. | Preclinical | Various solid cancer cell lines, osteosarcoma xenografts [142] | N/A | N/A |
| Sempervirine | MDM2 | MDM2–p53 inhibitor | Preclinical | Wt-p53 mouse embryonic fibroblast cell model [143] | N/A | N/A |
| RITA | MDM2 | Binds wt-p53, preventing association with MDM2 | Preclinical | Fibrosarcoma and colon cancer cell lines and wt-p53 tumor xenografts [144] | N/A | N/A |
| Syl-155 | MDM2 | Competitively inhibits MDM2–p53 binding | Preclinical | Wt-p53 fibrosarcoma cell line [145] | N/A | N/A |
| HLI373 | MDM2 | Competitively inhibits MDM2–p53 binding | Preclinical | Colon carcinoma cell line, wt-p53 transformed MEF cell model [146] | N/A | N/A |
| MEL24 | MDMX/MDM2 | Inhibits E3 ligase activity of MDM2–MDMX complex | Preclinical | Various wt-p53 cancer cell lines [147] | N/A | N/A |
| NSC207895 (XI-006) | MDMX | Represses MDMX transcription, activates p53 pathway | Preclinical | Various solid tumor cell lines [148] | N/A | N/A |
| CRL/SCF RING E3 inhibitors | ||||||
| Oridonin | FBW7 | Promotes proteasomal degradation of c-Myc via FBW7 agonism | Preclinical | Leukemia and lymphoma cell lines [149] | N/A | N/A |
| SCF-I2 | FBW7 | Blocks substrate-binding pocket, inhibits Cdc4, prevents substrate recognition | Preclinical | Colon and prostate cancer cell lines [150] | N/A | N/A |
| SMER3 | Met30 | Directly binds Met30, preventing association with SCF complex | Preclinical | N/A | N/A | N/A |
| Compound A | SKP2 | Prevents association of SKP2 with SCF complex, results in accumulation of p27 | Preclinical | MM cell lines, primary hematological malignancy cells [151] | N/A | N/A |
| SMIP004 | SKP2 | Downregulates SKP2, stabilizes p27 | Preclinical | Prostate adenocarcinoma cell lines [152] | N/A | N/A |
| C1, C2, C3 | SKP2 | Sterically inhibits SKP2–Cks1–p27 interface | Preclinical | Metastatic melanoma cell lines, breast cancer cells [153] | N/A | N/A |
| SZL-P1-41 (Compound #25) | SKP2 | Directly binds SKP2 to inhibit E3 activity | Preclinical | Prostate, lung, liver, and osteosarcoma tumor cell lines [154] | N/A | N/A |
| Longikaurin A | SKP2 | Downregulates SKP2 expression | Preclinical | Hepatocellular carcinoma [155] | N/A | N/A |
| Curcumin | SKP2 | Downregulates SKP2 | Phase 3 | Breast cancers [156], pancreatic cancer [157], glioma cells [158] | Prostate cancer, pancreatic cancer, colorectal cancer, MM, gastric cancer, breast cancer, leukemias and lymphomas, and more | [159,160] |
| Erioflorin | -TrCP1 | -TrCP1 to stabilize tumor suppressor Pdcd4 | Preclinical | Kidney, breast, ovarian, and colon cancer cell lines [161] | N/A | N/A |
| GS143 | -TrCP1 | -TrCP1 ubiquitination of IkB, suppresses NF-kB signaling | Preclinical | N/A | N/A | N/A |
| Other RING E3 inhibitors | ||||||
| CCW 28-3 | RNF4 | Binds RNF4 to facilitate degradation of BRD4 | Preclinical | Breast cancer cells [162] | N/A | N/A |
| GDC-0152 | IAPs | SMAC mimetic, induces IAP degradation, activates caspases | Preclinical (Phase 1 terminated) | Breast cancer cell lines and xenografts [163] | Solid cancers | N/A |
| LCL161 | IAPs | SMAC mimetic, induces degradation of cIAP-1 | Phase 2 | Various solid tumor cell lines [164], hepatocellular carcinoma [165], osteosarcoma [166], and more | Neoplasms, plasma cell myeloma, metastatic pancreatic cancer, myelofibrosis, small cell lung cancer, ovarian cancer, breast cancer, MM, and more | [167] |
| AT-406 (DEBIO1143, SM-406) | IAPs | SMAC mimetic; binds XIAP, cIAP-1, and cIAP-2; and activates caspases | Phase 3 | Breast cancer [168], colorectal cancer [169], ovarian cancer [170] | Lymphoma, solid tumors, AML, NSCLC, squamous cell carcinoma of head and neck, MM, and more | [171,172] |
| AEG 35,156 (GEM640), AEG 40826 | XIAP | Antisense oligonucleotides targeting XIAP mRNA to lower apoptotic threshold of cancer cells | Phase 2 | Various cancer cell lines, xenograft models of colon, breast and osteosarcoma tumors [173] | Advanced solid tumors, leukemia, mammary carcinoma, pancreatic carcinoma, BCL, NSCLC, hepatocellular carcinoma, and more | [174] |
| TL 32711 | IAPs | SMAC mimetic, induces degradation of cIAP-1, and caspase activation | Phase 2 | MM cell lines and animal models [175] | Chronic myelomonocytic leukemia, relapsed epithelial ovarian cancer, myelodysplastic syndrome, peritoneal neoplasms, and more | [176] |
| YM155 (sepantronium bromide) | IAPs | Inhibits promoter of survivin gene (IAP protein) | Phase 2 | Prostate cancer cell lines and xenografts [177] | Prostate cancer, melanoma, non-Hodgkin’s lymphoma, breast cancer, diffuse large-cell lymphoma, refractory B-cell lymphoma, and more | [178] |
| C25-140 | TRAF6 | Inhibits TRAF6–Ubc13 interaction specifically | Preclinical | Only studied in autoimmune and inflammatory disease models [179] | N/A | N/A |
| BC-1215 | TRAF6 (via FBXO3 inhibition) | Antagonist of FBXO3, destabilizes TRAF6 | Preclinical | Only studied in autoimmune and inflammatory disease models [180,181] | N/A | N/A |
| HECT E3 ligase inhibitors | ||||||
| HS-152 | SMURF1 | Reversibly blocks SMURF1-mediated RHOB ubiquitination | Preclinical | Inhibited protrusive RHOB-dependent activity in cell lines [182] | N/A | N/A |
| BI8622 and BI8626 | HUWE1 | Inhibit HUWE1 to stabilize assembly of Myc-repressive MIZ1 complex on Myc-activated target genes | Preclinical | Colorectal cancer [183], MM [184] | N/A | N/A |
| Compound 12 | E6AP | Inhibits oncogenic E6–p53 interaction in E6–E6AP–p53 complex | Preclinical | HPV-positive cervical carcinoma cell lines [185] | N/A | N/A |
| Lutolein and CAF024 | E6AP | Bind viral E6 to prevent hijacking of E6AP | Preclinical | HPV-positive cervical carcinoma cells [186] | N/A | N/A |
| Lig1, Lig2, Lig3 | E6AP | Inhibit E6–E6AP interaction | Preclinical | N/A | N/A | N/A |
| N-acetyl phenylalanine | E6AP | Dissociates active E6AP trimer | Preclinical | N/A | N/A | N/A |
| CM11-1 | E6AP | E6AP inhibitor, prevents polyubiquitination of Prx1 in E6-independent and -dependent manner | Preclinical | N/A | N/A | N/A |
| Heclin | HECT, non-specific | Induces conformational change in HECT domain to inhibit activity | Preclinical | N/A | N/A | N/A |
| RBR E3 ligase inhibitors | ||||||
| BAY 11-7082 | LUBAC | Covalently binds active cysteine residues of E2′s Ubc13 and UbcH7 to prevent Ub conjugation | Preclinical | B-cell lymphoma, leukemia [187], gastric cancer [188] | N/A | N/A |
| gliotoxin | LUBAC | Selectively binds RBR domain of HOIP to inhibit Ub chain formation | Preclinical | N/A | N/A | N/A |
| HOIPIN-8 | LUBAC | Inhibits LUBAC activity and suppresses NF-kB activation | Preclinical | ABC-DLBCL [189,190] | N/A | N/A |
| Bendamustine | LUBAC | Specifically inhibits HOIP | FDA approved (Phase 4) | Chronic lymphocytic leukemia, MM, non-Hodgkin’s lymphoma [191], and more | Ovarian cancer, MM, relapsed T-cell lymphoma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, and more | [192,193,194,195] |
| Compound Name | UPS Target | Mode of Action | Current Clinical Stage | Cancer Models Targeted in Preclinical Studies | Clinical Trial Cancer Targets |
|---|---|---|---|---|---|
| HBX 41108, HBX 19818, HBX 28258 | USP7 | Covalently binds USP7 active site, increasing p53 | Preclinical | Colon cancer cell lines [249,252] | N/A |
| P5091 | USP7 and USP47 | Dual inhibitor of USP7 and USP47 | Preclinical | MM cells, bortezomib-resistant MM, and B-cell leukemia mouse models [253] | N/A |
| P50429, P22077 | USP7 | Irreversibly bind catalytic cysteine residue of USP7 | Preclinical | Colon cancer cell line [254], neuroblastoma xenograft [255] | N/A |
| Compound 4 | USP7 | Selectively binds USP7 allosteric site | Preclinical | Leukemia, prostate adenocarcinoma cell lines [259] | N/A |
| GNE-6640, GNE-6776 | USP7 | Binds novel USP7 functional site | Preclinical | Colon cancer cells [260] | N/A |
| XL188 | USP7 | Non-covalent inhibition of USP7 active site, causing accumulation of p53 and p21 | Preclinical | Breast cancer and MM cell lines [261] | N/A |
| FT671, FT827 | USP7 | Allosteric USP7 inhibition | Preclinical | Colon cancer, osteosarcoma cell lines, MM cells, and xenografts [262] | N/A |
| Pimozide and GW7647 | USP1/UAP1 complex | Non-competitive inhibition of USP1 | Preclinical | NSCLC cells [263] | N/A |
| IU1, IU1-47, 1B10, IU1-248 | USP14 | Binds USP14 steric site | Preclinical | N/A | N/A |
| b-AP15 | USP14 and UCHL5 | Blocks 19S DUB activity | Preclinical | Solid tumors, MM [40,72,73,74] | N/A |
| VLX1570 | USP14 and UCHL5 | b-AP15 analog, also blocks 19S DUBs | Preclinical (Phase 1/2 terminated) | MM [42] | Multiple Myeloma (MM) |
| Capzimin, 8TQ | Rpn11 | Specific inhibitors of Rpn11 | Preclinical | Leukemia, NSCLC, breast cancer [79] | N/A |
| Thiolutin, SOP11 | Rpn11 (nonspecific) | Non-specifically inhibits JAMM-family DUBs | Preclinical | Colon cancer, bortezomib-resistant RPE cells [80] | N/A |
| Celasterol, mangaferin | UCHL1 | Reversible inhibitors of UCHL1 | Preclinical | N/A | N/A |
| LDN-57444, LDN-Pox | UCHL1 | Incorporation of LDN-57444 in Pox micelle delivery system inhibits UCHL1 in cells | Preclinical | Oral squamous cell carcinoma cell line [264] | N/A |
| 6RK73 and 8RK64 | UCHL1 | Cell penetrable probes bind UCHL1 active site cysteine in activity-dependent manner | Preclinical | N/A | N/A |
| WP1130 | USP9X | Inhibits USP9X, UCHL, and USP14 to a degree | Preclinical | CML, melanoma, glioblastoma, myeloproliferative disorders [44,75,76], MCL [77], AML [78] | N/A |
| EOAI3402143 (G9) | USP9X/USP24 (and partially USP5) | WP1130 analog | Preclinical | AML [78], MM [265], melanoma [76] | N/A |
| FT709 | USP9X | Competitively binds USP9X active site | Preclinical | Colon cancer cell line [266] | N/A |
| OTUB2-COV-1 | OTUB2 | Covalent OTUB2 inhibitor | Preclinical | N/A | N/A |
| BC-1471 | STAMBP | Inhibits STAMBP, prevents interaction with NALP7 | Preclinical | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LaPlante, G.; Zhang, W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers 2021, 13, 3079. https://doi.org/10.3390/cancers13123079
LaPlante G, Zhang W. Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers. 2021; 13(12):3079. https://doi.org/10.3390/cancers13123079
Chicago/Turabian StyleLaPlante, Gabriel, and Wei Zhang. 2021. "Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors" Cancers 13, no. 12: 3079. https://doi.org/10.3390/cancers13123079
APA StyleLaPlante, G., & Zhang, W. (2021). Targeting the Ubiquitin-Proteasome System for Cancer Therapeutics by Small-Molecule Inhibitors. Cancers, 13(12), 3079. https://doi.org/10.3390/cancers13123079