
Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
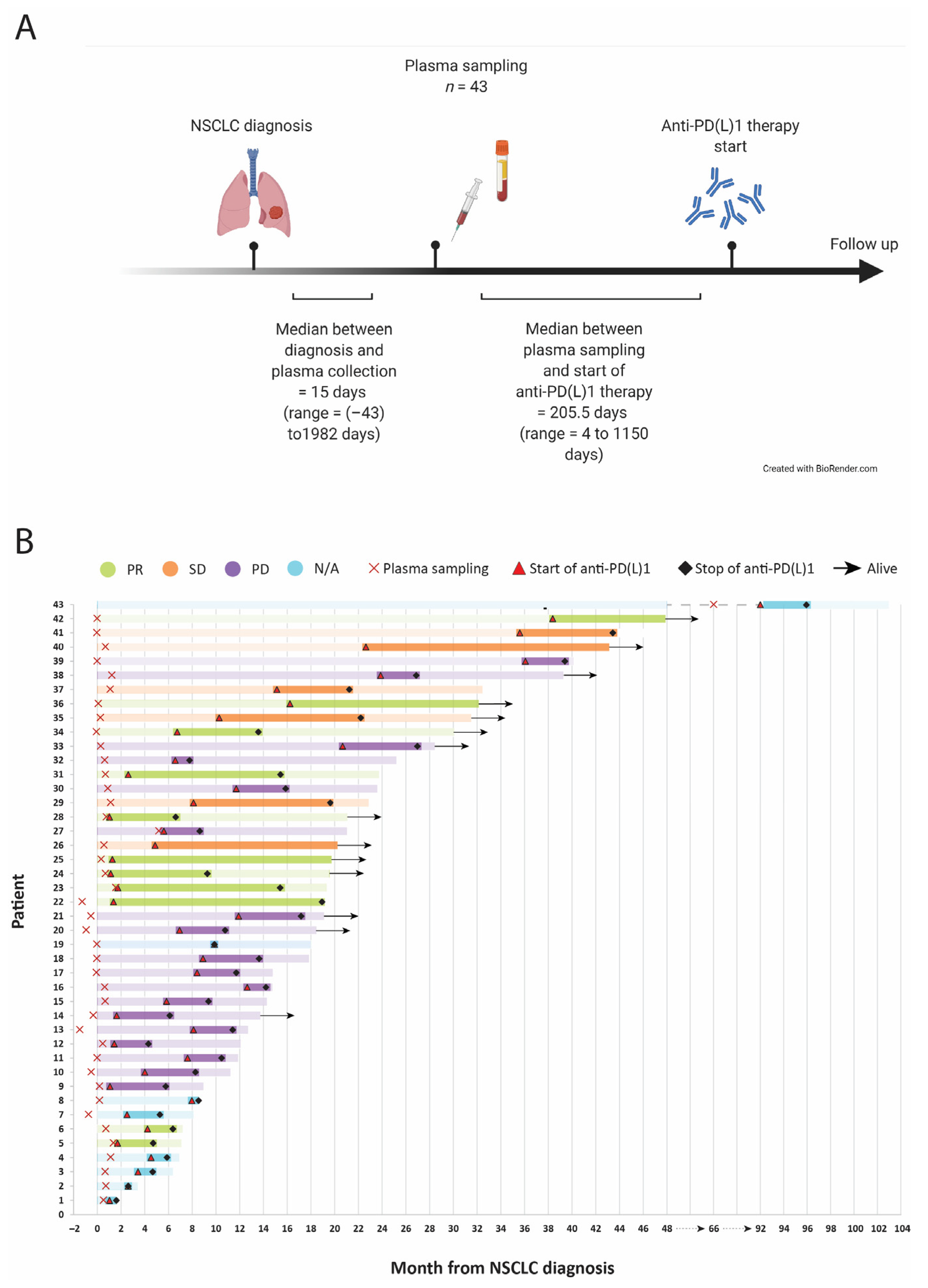
2.1. Patient Cohorts and Samples
2.2. Sample Analysis
2.3. Statistical Analysis
2.4. Ethical Considerations
3. Results
3.1. Patient Cohorts Clinical and Sample Characteristics
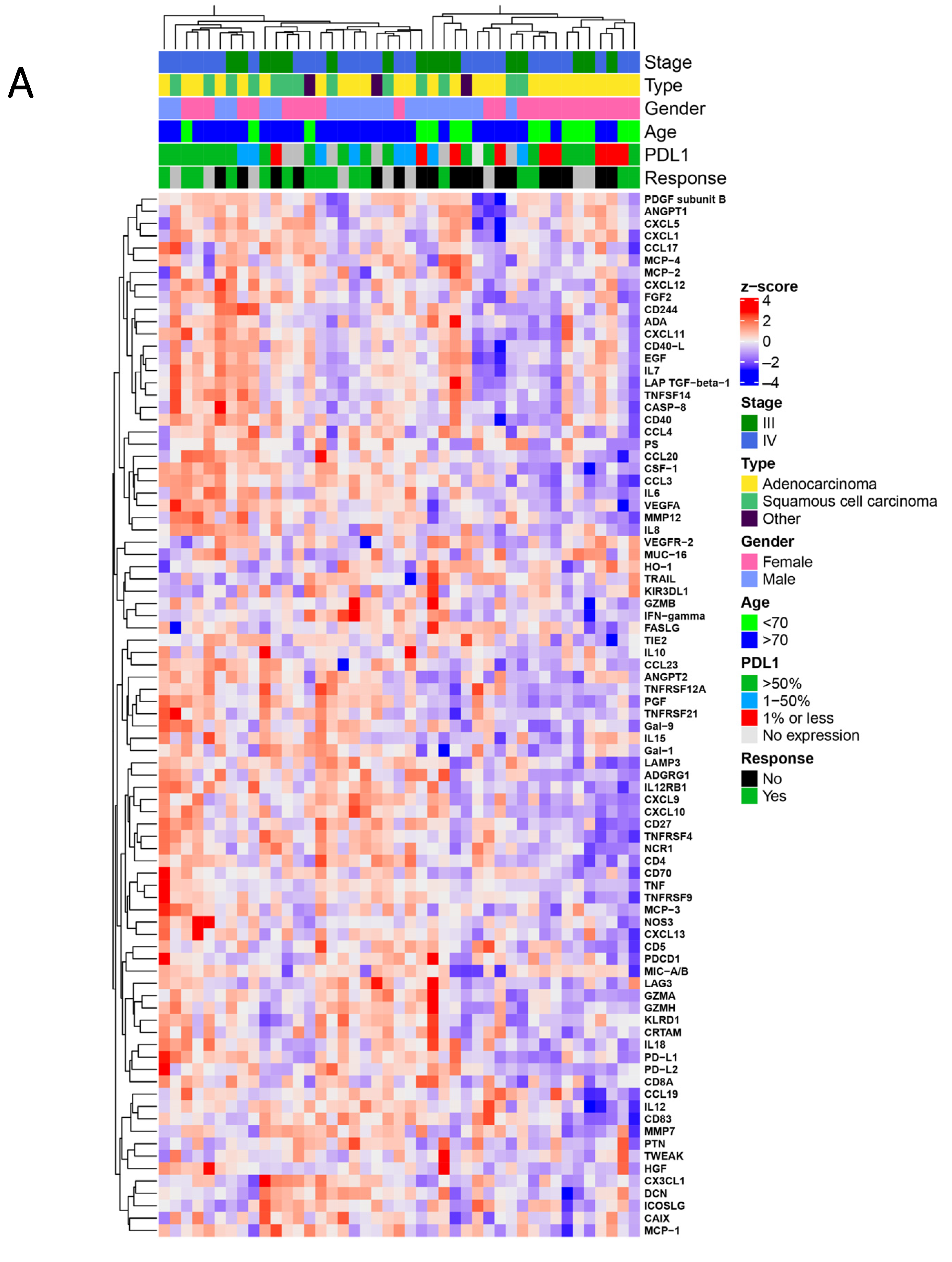
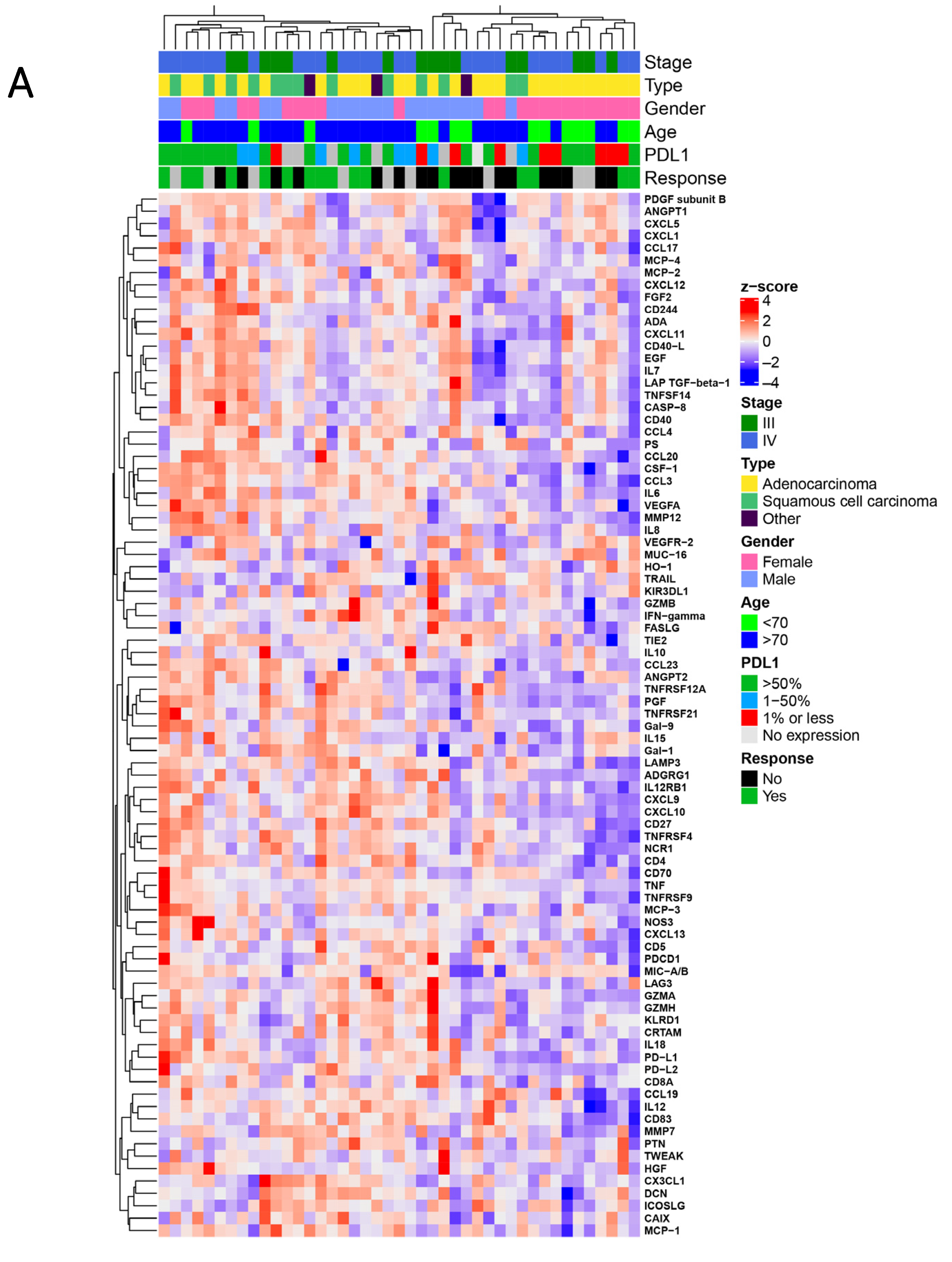
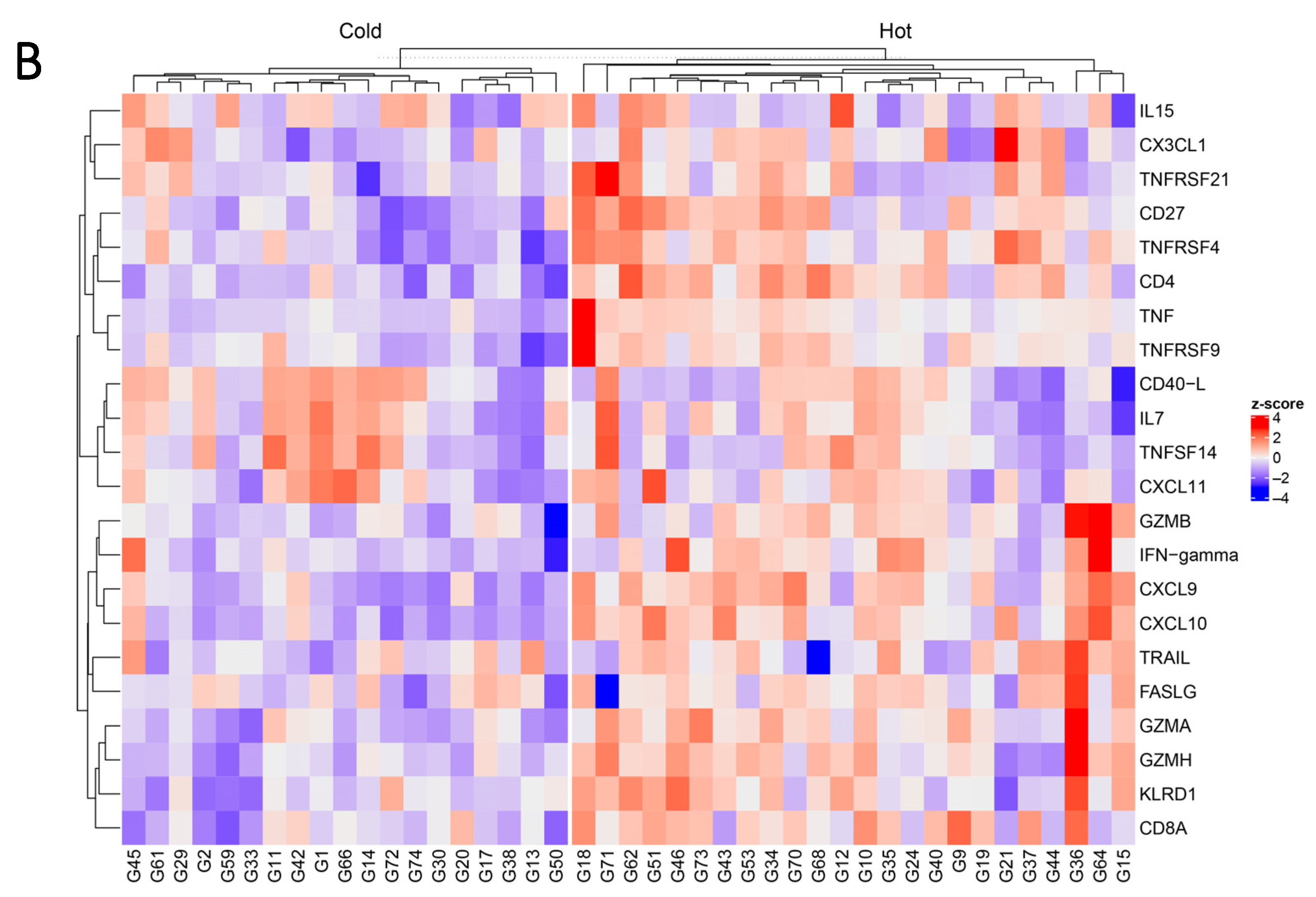
3.2. Proteomic Signatures Detected in the Plasma of NSCLC Patients
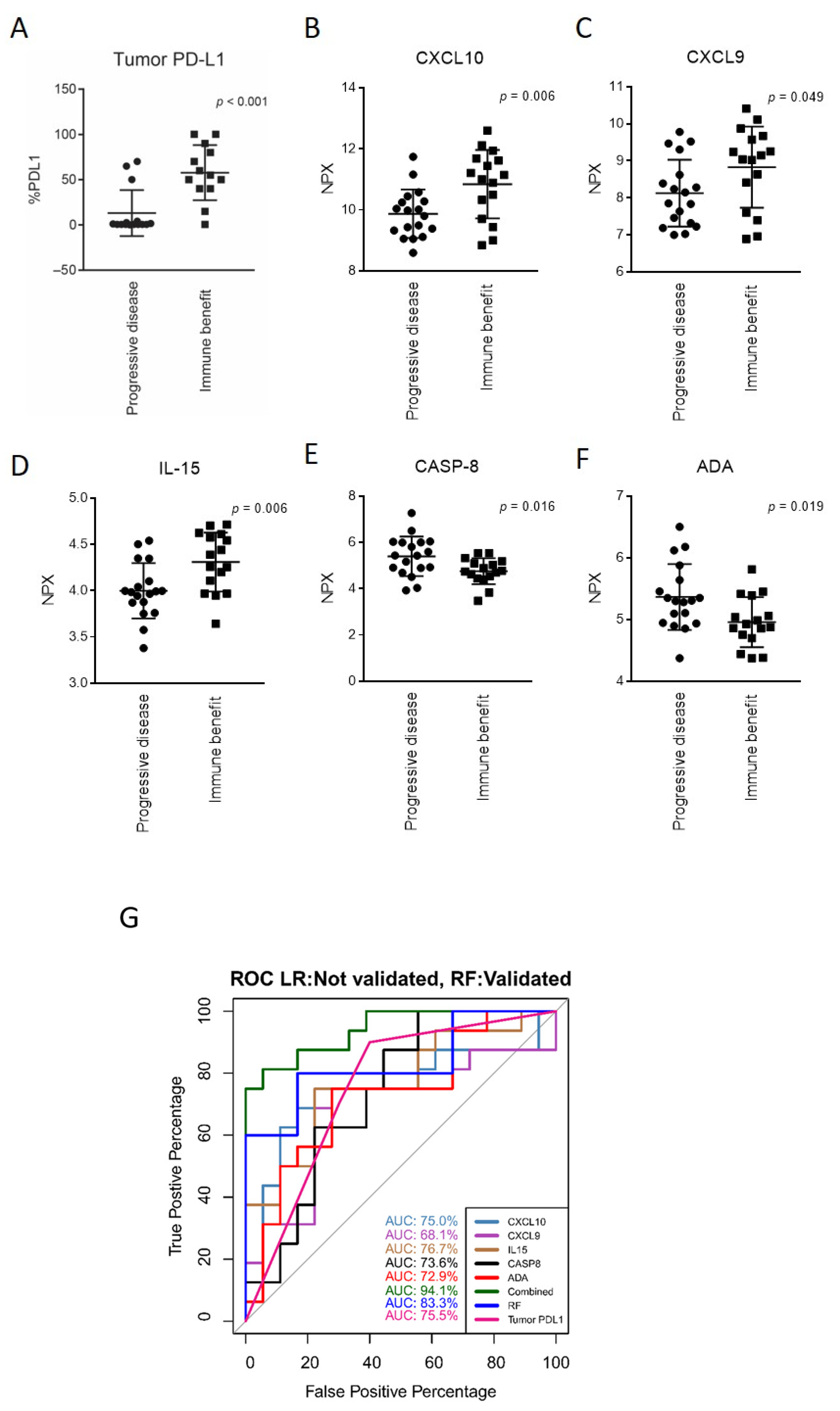
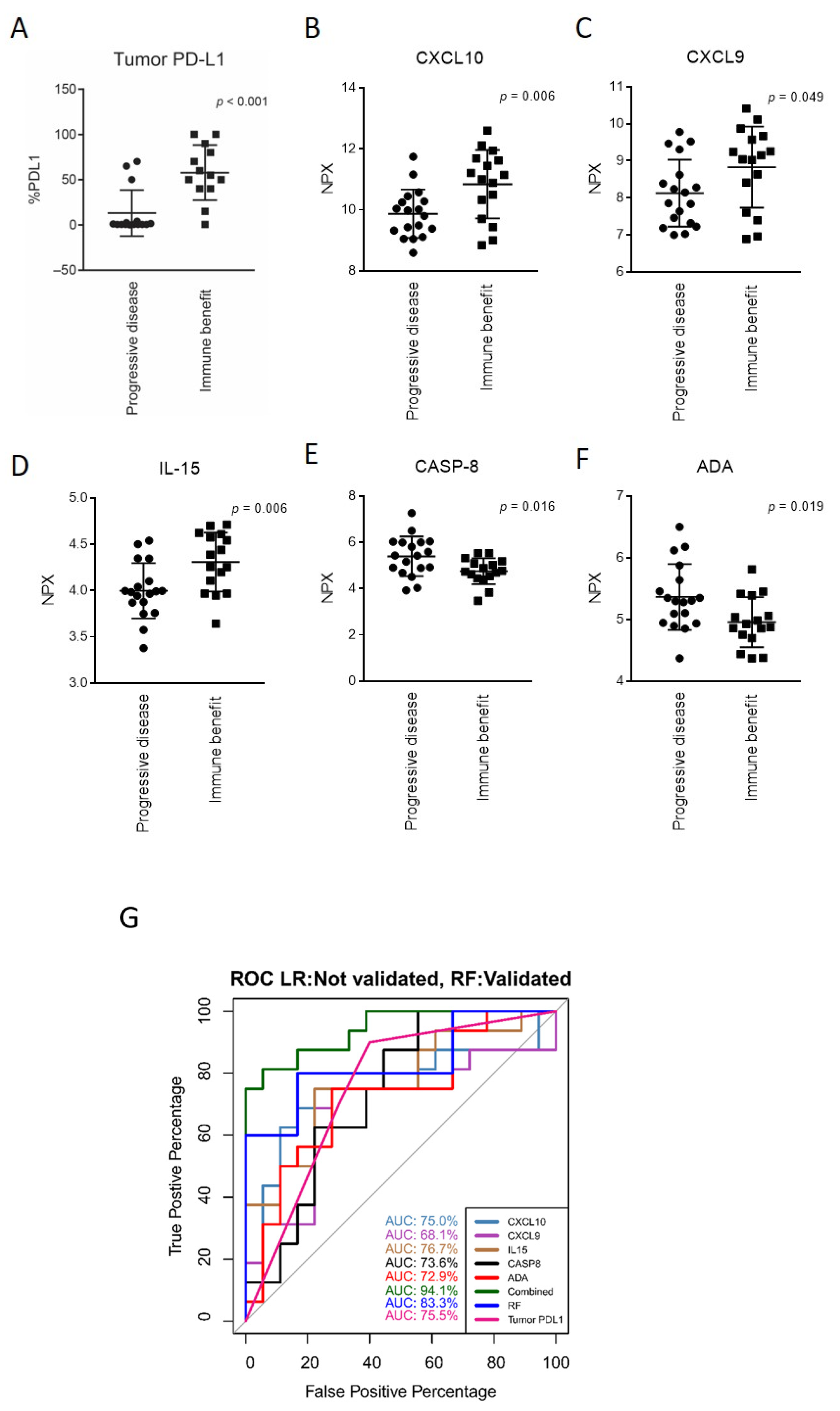
3.3. Markers of Response to Anti-PD-(L)1 Therapy and a Response-Predictive Signature
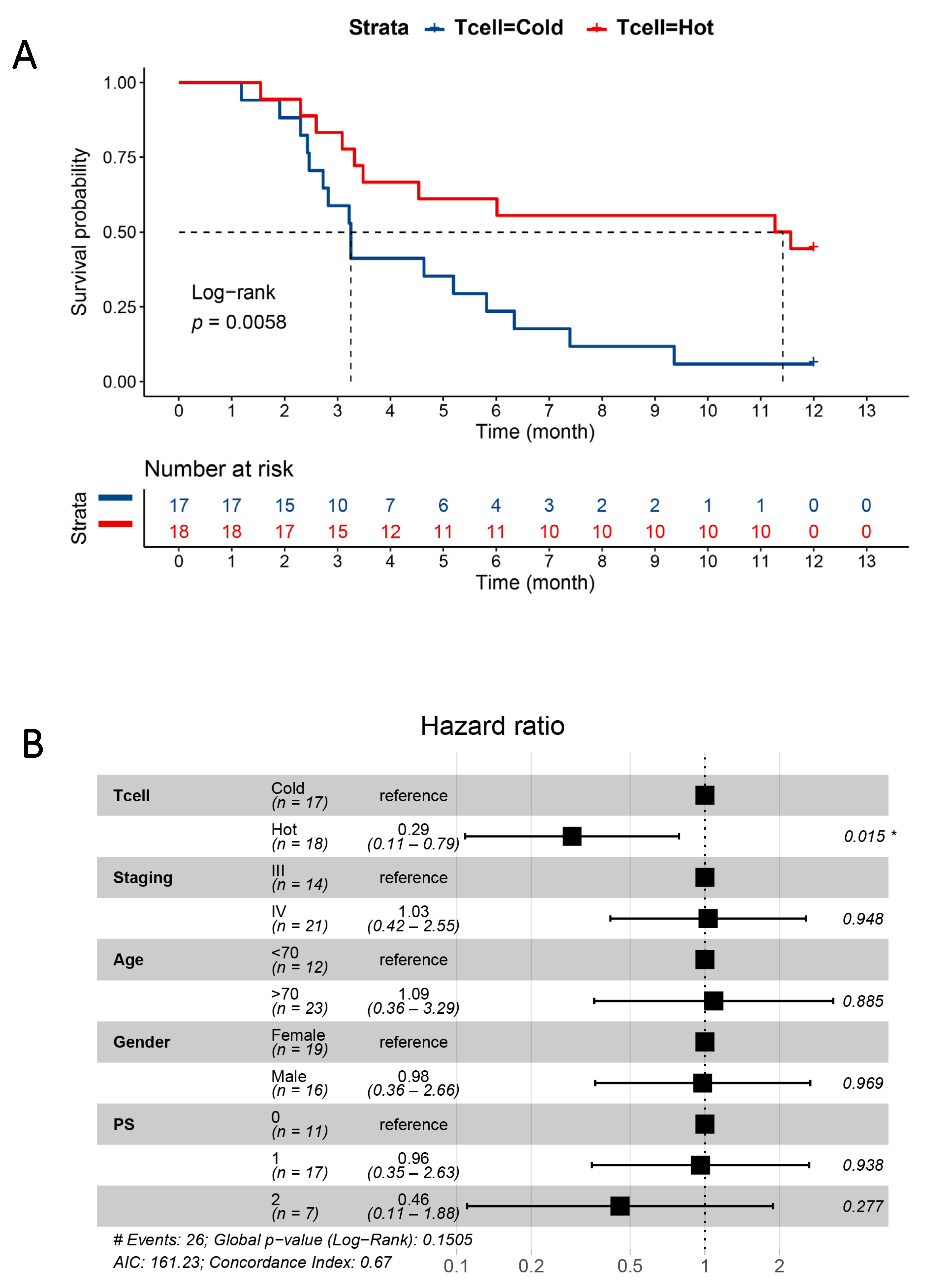
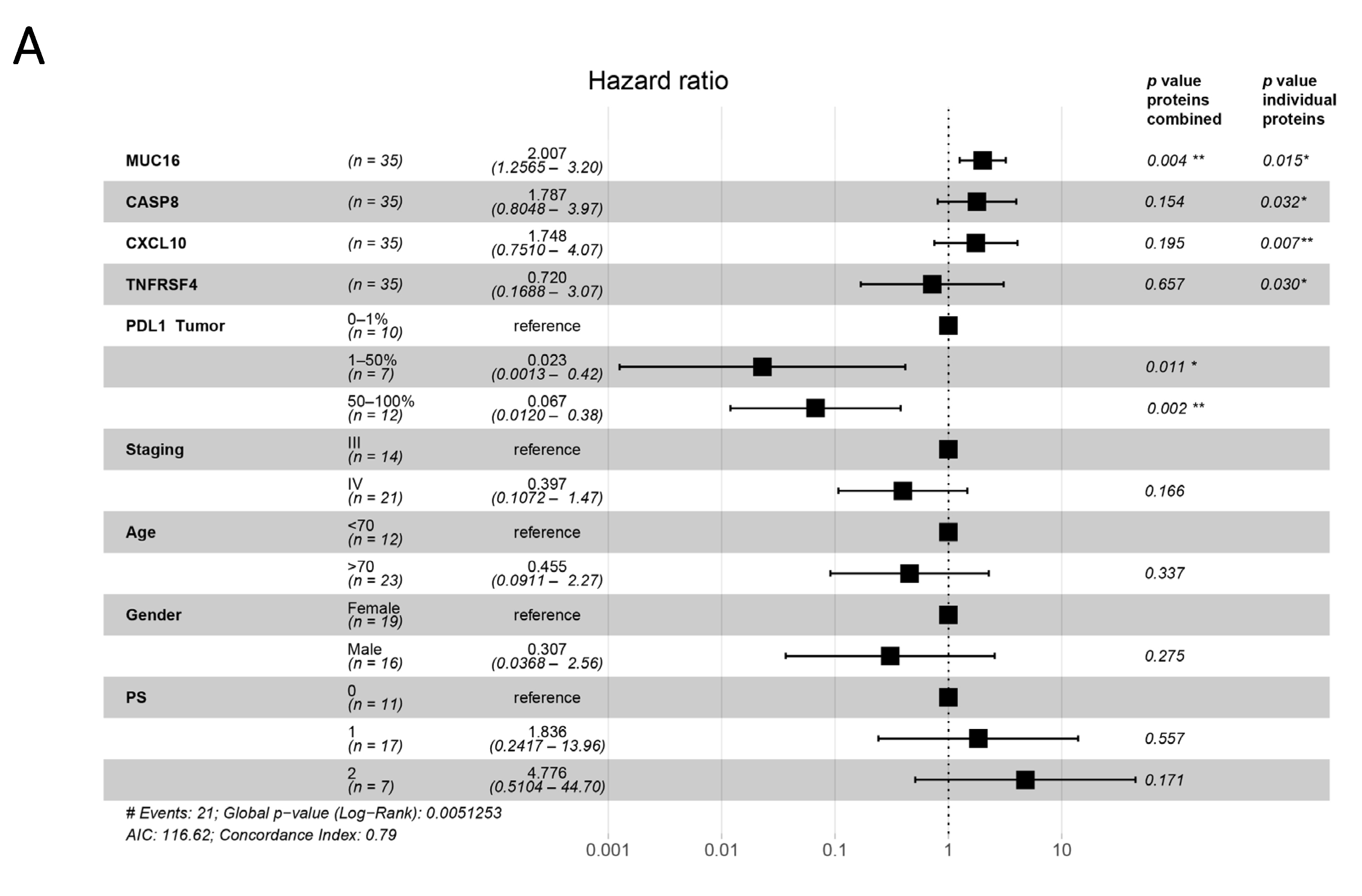
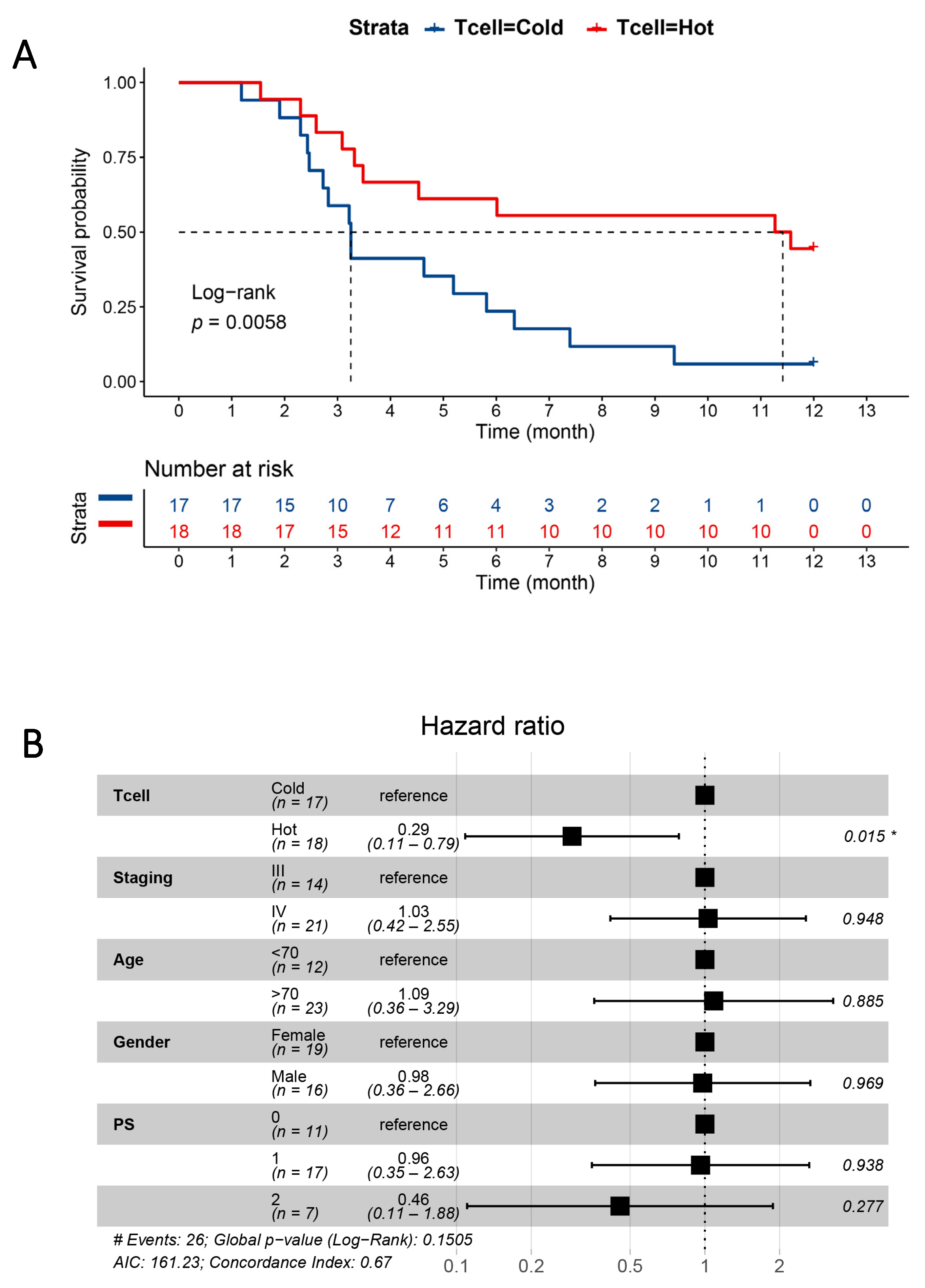
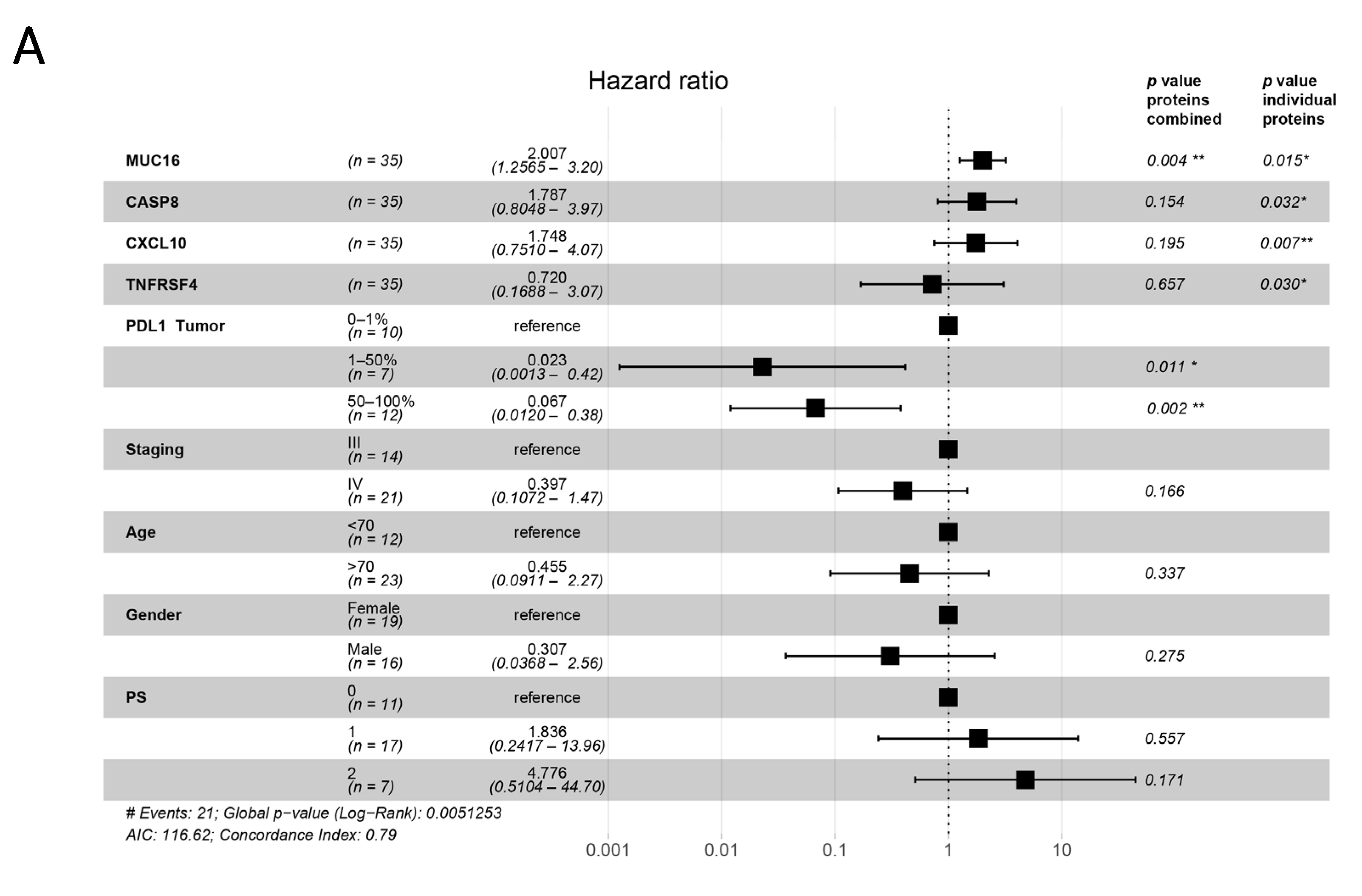
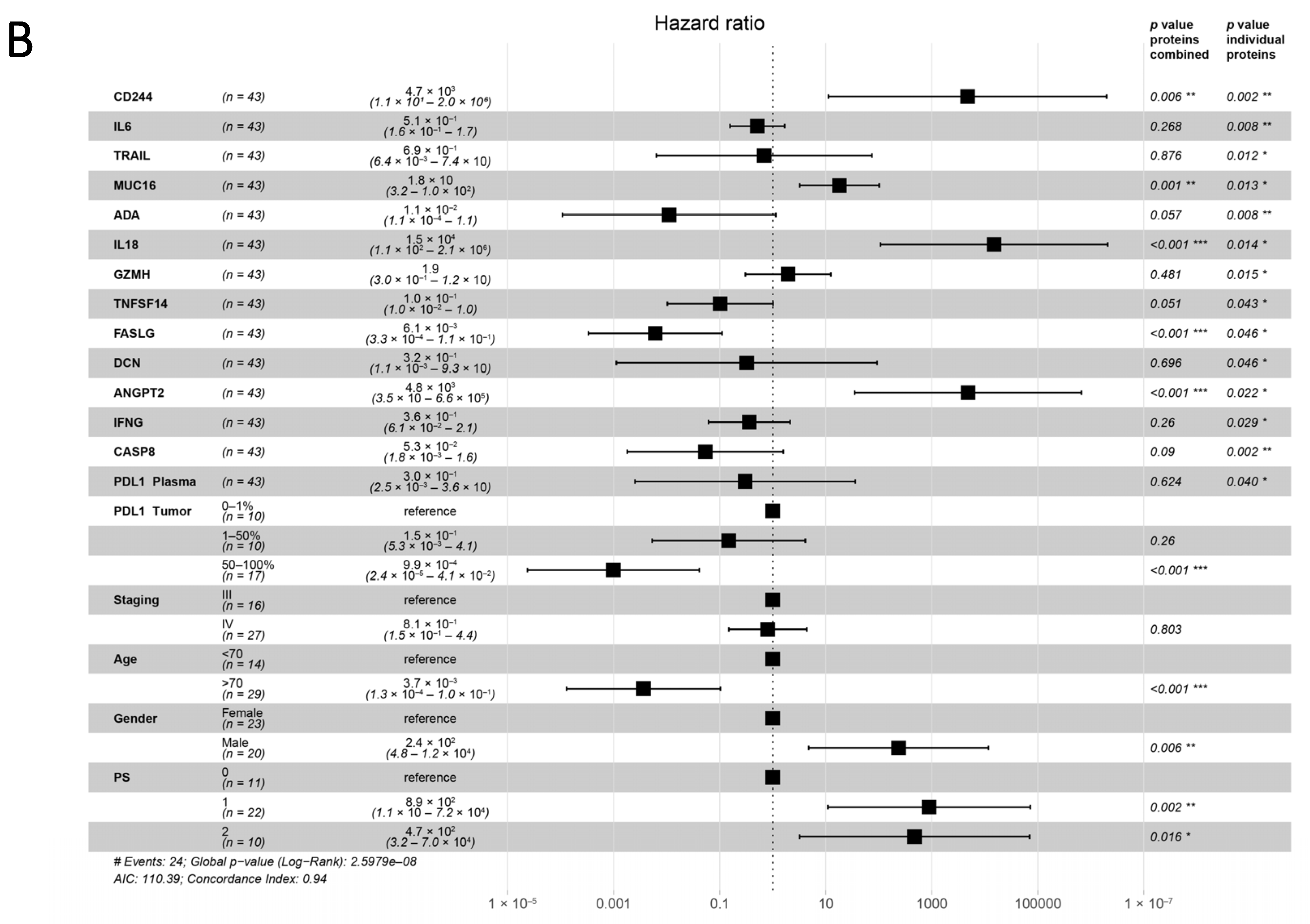
3.4. Plasma Proteins as Survival Predictors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Perez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Wagner, G.; Stollenwerk, H.K.; Klerings, I.; Pecherstorfer, M.; Gartlehner, G.; Singer, J. Efficacy and safety of immune checkpoint inhibitors in patients with advanced non–small cell lung cancer (NSCLC): A systematic literature review. OncoImmunology 2020, 9, 1774314. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Hersom, M.; Jørgensen, J.T. Companion and Complementary Diagnostics–Focus on PD-L1 Expression Assays for PD-1/PD-L1 Checkpoint Inhibitors in Non–Small Cell Lung Cancer. Ther. Drug Monit. 2018, 40, 9–16. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Tsao, M.; Kerr, K.M.; Kockx, M.; Beasley, M.-B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.-Y.; et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Brunnström, H.; Johansson, A.; Westbom-Fremer, S.; Backman, M.; Djureinovic, D.; Patthey, A.; Isaksson-Mettävainio, M.; Gulyas, M.; Micke, P. PD-L1 immunohistochemistry in clinical diagnostics of lung cancer: Inter-pathologist variability is higher than assay variability. Mod. Pathol. 2017, 30, 1411–1421. [Google Scholar] [CrossRef] [Green Version]
- Elfving, H.; Mattsson, J.S.M.; Lindskog, C.; Backman, M.; Menzel, U.; Micke, P. Programmed Cell Death Ligand 1 Immunohisto-chemistry: A Concordance Study Between Surgical Specimen, Biopsy, and Tissue Microarray. Clin. Lung Cancer 2019, 20, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Sesma, A.; Pardo, J.; Cruellas, M.; Gálvez, E.M.; Gascón, M.; Isla, D.; Martínez-Lostao, L.; Ocáriz, M.; Paño, J.R.; Quílez, E.; et al. From Tumor Mutational Burden to Blood T Cell Receptor: Looking for the Best Predictive Biomarker in Lung Cancer Treated with Immunotherapy. Cancers 2020, 12, 2974. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Stenzinger, A.; Peters, S. Tumor Mutational Burden as a Pan-cancer Biomarker for Immunotherapy: The Limits and Potential for Convergence. Cancer Cell 2020, 38, 624–625. [Google Scholar] [CrossRef] [PubMed]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indini, A.; Rijavec, E.; Grossi, F. Circulating Biomarkers of Response and Toxicity of Immunotherapy in Advanced Non-Small Cell Lung Cancer (NSCLC): A Comprehensive Review. Cancers 2021, 13, 1794. [Google Scholar] [CrossRef] [PubMed]
- Soyano, A.E.; Dholaria, B.; Marin-Acevedo, J.A.; Diehl, N.; Hodge, D.; Luo, Y.; Manochakian, R.; Chumsri, S.; Adjei, A.; Knutson, K.L.; et al. Peripheral blood biomarkers correlate with out-comes in advanced non-small cell lung Cancer patients treated with anti-PD-1 antibodies. J. Immunother. Cancer 2018, 6, 129. [Google Scholar] [CrossRef]
- Okuma, Y.; Wakui, H.; Utsumi, H.; Sagawa, Y.; Hosomi, Y.; Kuwano, K.; Homma, S. Soluble Programmed Cell Death Ligand 1 as a Novel Biomarker for Nivolumab Therapy for Non–Small-cell Lung Cancer. Clin. Lung Cancer 2018, 19, 410–417.e1. [Google Scholar] [CrossRef]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Incorvaia, L.; Fanale, D.; Badalamenti, G.; Porta, C.; Olive, D.; De Luca, I.; Brando, C.; Rizzo, M.; Messina, C.; Rediti, M.; et al. Baseline plasma levels of soluble PD-1, PD-L1, and BTN3A1 predict response to nivolumab treatment in patients with metastatic renal cell carcinoma: A step toward a biomarker for therapeutic decisions. OncoImmunology 2020, 9, 1832348. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Park, C.-K.; Chung, C.; Oh, I.-J.; Kim, Y.-C.; Park, D.; Kim, J.; Kwon, G.C.; Kwon, I.; Sun, P.; et al. Baseline Serum Interleukin-6 Levels Predict the Response of Patients with Advanced Non-small Cell Lung Cancer to PD-1/PD-L1 Inhibitors. Immune Netw. 2020, 20, e27. [Google Scholar] [CrossRef] [PubMed]
- Costantini, A.; Julie, C.; Dumenil, C.; Hélias-Rodzewicz, Z.; Tisserand, J.; Dumoulin, J.; Giraud, V.; Labrune, S.; Chinet, T.; Emile, J.-F.; et al. Predictive role of plasmatic biomarkers in advanced non-small cell lung cancer treated by nivolumab. OncoImmunology 2018, 7, e1452581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.; Badgery-Parker, T.; Dobbins, T.; Jorgensen, M.; Gibbs, P.; Faragher, I.; Jones, I.; Currow, D. Comparison of ECOG/WHO Performance Status and ASA Score as a Measure of Functional Status. J. Pain Symptom Manag. 2015, 49, 258–264. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Augustus, E.; Zwaenepoel, K.; Siozopoulou, V.; Raskin, J.; Jordaens, S.; Baggerman, G.; Sorber, L.; Roeyen, G.; Peeters, M.; Pauwels, P. Prognostic and Predictive Biomarkers in Non-Small Cell Lung Cancer Patients on Immunotherapy—The Role of Liquid Biopsy in Unraveling the Puzzle. Cancers 2021, 13, 1675. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef] [Green Version]
- Veldore, V.H.; Choughule, A.; Routhu, T.; Mandloi, N.; Noronha, V.; Joshi, A.; Dutt, A.; Gupta, R.; Vedam, R.; Prabhash, K. Validation of liquid biopsy: Plasma cell-free DNA testing in clinical management of advanced non-small cell lung cancer. Lung Cancer Targets Ther. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djureinovic, D.; Pontén, V.; Landelius, P.; Al Sayegh, S.; Kappert, K.; Kamali-Moghaddam, M.; Micke, P.; Ståhle, E. Multiplex plasma protein profiling identifies novel markers to discriminate patients with adenocarcinoma of the lung. BMC Cancer 2019, 19, 741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottet, N.; Bellmunt, J.; Bolla, M.; Briers, E.; Cumberbatch, M.G.; De Santis, M.; Fossati, N.; Gross, T.; Henry, A.; Joniau, S.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2017, 71, 618–629. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunology 2013, 39, 782–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farber, J.M. A macrophage mRNA selectively induced by γ-interferon encodes a member of the platelet factor 4 family of cytokines. Proc. Natl. Acad. Sci. USA 1990, 87, 5238–5242. [Google Scholar] [CrossRef] [Green Version]
- Luster, A.D.; Unkeless, J.C.; Ravetch, J.V. γ-Interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nat. Cell Biol. 1985, 315, 672–676. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 64, 40–47. [Google Scholar] [CrossRef]
- Gorbachev, A.V.; Kobayashi, H.; Kudo, D.; Tannenbaum, C.S.; Finke, J.H.; Shu, S.; Farber, J.M.; Fairchild, R.L. CXC Chemokine Ligand 9/Monokine Induced by IFN-γ Production by Tumor Cells Is Critical for T Cell-Mediated Suppression of Cutaneous Tumors. J. Immunol. 2007, 178, 2278–2286. [Google Scholar] [CrossRef] [Green Version]
- Arenberg, D.A.; White, E.; Burdick, M.D.; Strom, S.R.; Strieter, R.M. Improved survival in tumor-bearing SCID mice treated with interferon-γ-inducible protein 10 (IP-10/CXCL10). Cancer Immunol. Immunother. 2001, 50, 533–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissler, N.; Mao, Y.; Brodin, D.; Reuterswärd, P.; Svahn, H.A.; Johnsen, J.I.; Kiessling, R.; Kogner, P. Regulation of myeloid cells by activated T cells determines the efficacy of PD-1 blockade. OncoImmunology 2016, 5, e1232222. [Google Scholar] [CrossRef] [Green Version]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudson, K.M.; Hicks, K.C.; Alter, S.; Schlom, J.; Gameiro, S.R. Mechanisms involved in IL-15 superagonist enhancement of an-ti-PD-L1 therapy. J. Immunother. Cancer. 2019, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Stupack, D.G. Caspase-8 as a therapeutic target in cancer. Cancer Lett. 2013, 332, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Colucci, R.L.; La Motta, C.; Tuccori, M.; Awwad, O.; Da Settimo, F.; Blandizzi, C.; Fornai, M. Adenosine Deaminase in the Modulation of Immune System and its Potential as a Novel Target for Treatment of Inflammatory Disorders. Curr. Drug Targets 2012, 13, 842–862. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Karami-Tehrani, F.; Salami, S.; Atri, M. Adenosine deaminase activity in the serum and malignant tumors of breast cancer: The assessment of isoenzyme ADA1 and ADA2 activities. Clin. Biochem. 2005, 38, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 2020, 181, 674–687.e13. [Google Scholar] [CrossRef]
- Morello, S.; Pinto, A.; Blandizzi, C.; Antonioli, L. Myeloid cells in the tumor microenvironment: Role of adenosine. OncoImmunology 2015, 5, e1108515. [Google Scholar] [CrossRef] [Green Version]
- Meyo, M.T.; Jouinot, A.; Giroux-Leprieur, E.; Fabre, E.; Wislez, M.; Alifano, M.; Leroy, K.; Boudou-Rouquette, P.; Tlemsani, C.; Khoudour, N.; et al. Predictive Value of Soluble PD-1, PD-L1, VEGFA, CD40 Ligand and CD44 for Nivolumab Therapy in Advanced Non-Small Cell Lung Cancer: A Case-Control Study. Cancers 2020, 12, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; E Rodriguez-Ruiz, M.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristic | n (%) |
|---|---|
| All patients | 43 (100) |
| Sex | |
| Female | 23 (53.5) |
| Male | 20 (46.5) |
| Median age (years; at the start of immunotherapy) | 72.5 ± 8.8 |
| Smoking status (at diagnosis) | |
| Ever smoker (smoker/ex-smoker) | 39 (90.7) |
| Never smoker | 4 (9.3) |
| Tumor type | |
| Adenocarcinoma | 27 (62.8) |
| Squamous cell carcinoma | 13 (30.2) |
| Other | 3 (7.0) |
| Stage | |
| III | 16 (37.2) |
| IV | 27 (62.8) |
| Performance status | |
| 0 1 2 | 11 (25.6) 22 (51.2) 10 (23.2) |
| PD-L1 | |
| No expression | 1 (2.3) |
| <1% | 9 (20.9) |
| 1–49% 50–100% | 8 (18.6) 19 (44.2) |
| N/A | 6 (14.0) |
| Therapeutic agent | |
| Atezolizumab | 7 (16.3) |
| Durvalumab | 1 (2.3) |
| Nivolumab | 12 (27.9) |
| Pembrolizumab | 23 (53.5) |
| Therapy line at which immunotherapy was initiated | |
| 1st line | 13 (30.2) |
| 2nd line | 19 (44.2) |
| 3rd line | 9 (20.9) |
| 4th line | 1 (2.3) |
| 5th line | 1 (2.3) |
| Response | |
| Progressive disease (PD) | 18 (41.9) |
| Stable disease (SD) | 6 (14.0) |
| Partial response (PR) | 11 (25.6) |
| Complete remission (CR) | 0 (0.0) |
| N/A | 8 (18.5) |
| Clinical benefit (SD, PR) Progressive disease (PD) N/A | 17 (39.6) 18 (41.9) 8 (18.5) |
| Survival (from the start of immunotherapy) | |
| Median overall survival (days) ± SD | 315 ± 198.1 |
| Median progression-free survival (days) ± SD | 141 ± 202.5 |
| Clinical Characteristic | Immune “Hot” | Immune “Cold” | p-Value |
|---|---|---|---|
| Protein Signature | Protein Signature | ||
| n = 24 (55.8%) | n = 19 (44.2%) | ||
| Sex | 0.03 | ||
| Female | 9 (37.5) | 14 (73.7) | |
| Male | 15 (62.5) | 5 (26.3) | |
| Median age (years; at the start of immunotherapy) | 74.5 | 68.0 | 0.005 |
| Smoking status (at diagnosis) | >0.999 | ||
| Ever smoker (smoker/ex-smoker) | 22 (91.7) | 17 (89.5) | |
| Never smoker | 2 (8.3) | 2 (10.5) | |
| Tumor type | 0.674 | ||
| Adenocarcinoma | 15 (62.5) | 12 (63.2) | |
| Squamous cell carcinoma | 8 (33.3) | 5 (26.3) | |
| Other | 1 (4.2) | 2 (10.5) | |
| Stage | 0.752 | ||
| III | 6 (31.6) | 10 (41.7) | |
| IV | 13 (68.4) | 14 (58.3) | |
| Performance status | 0.203 | ||
| 0 1 2 | 4 (16.7) 15 (62.5) 5 (20.8) | 7 (36.8) 7 (36.8) 5 (26.3) | |
| PD-L1 | 0.197 | ||
| No expression | 0 (0.0) | 0 (0.0) | |
| <1% | 3 (12.5) | 7 (36.8) | |
| 1–49% 50–100% | 6 (25.0) 11 (45.8) | 4 (21.1) 6 (31.6) | |
| N/A | 4 (15.8) | 2 (10.5) | |
| Therapeutic agent | 0.643 | ||
| Atezolizumab | 3 (12.5) | 4 (21.1) | |
| Durvalumab | 1 (4.2) | 0 (0.0) | |
| Nivolumab | 6 (25.0) | 6 (31.6) | |
| Pembrolizumab | 14 (58.3) | 9 (47.4) | |
| Therapy line at which immunotherapy was initiated | 0.246 | ||
| 1st line | 10 (41.7) | 3 (15.8) | |
| 2nd line | 9 (37.5) | 10 (52.6) | |
| 3rd line | 4 (16.7) | 5 (26.3) | |
| 4th line | 1 (4.2) | 0 (0.0) | |
| 5th line | 0 (0.0) | 1 (5.3) | |
| Response | 0.065 | ||
| Progressive disease (PD) | 6 (25.0) | 12 (63.2) | |
| Stable disease (SD) | 5 (20.8) | 1 (5.3) | |
| Partial response (PR) | 7 (29.2) | 4 (21.1) | |
| Complete remission (CR) | 0 (0.0) | 0 (0.0) | |
| N/A | 6 (33.3) | 2 (10.5) | |
| Clinical benefit (SD, PR, CR) progressive disease (PD) N/A | 12 (50.0) 6 (25.0) 6 (25.0) | 4 (21.1) 12 (63.2) 3 (15.8) | 0.020 |
| Survival (from the start of immunotherapy) | |||
| Median overall survival (days) ± SD | 402.5 ± 213.0 | 236.0 ± 178.8 | - |
| Median progression-free survival (days) ± SD | 106.0 ± 125.9 | 98.5 ± 56.0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eltahir, M.; Isaksson, J.; Mattsson, J.S.M.; Kärre, K.; Botling, J.; Lord, M.; Mangsbo, S.M.; Micke, P. Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade. Cancers 2021, 13, 3116. https://doi.org/10.3390/cancers13133116
Eltahir M, Isaksson J, Mattsson JSM, Kärre K, Botling J, Lord M, Mangsbo SM, Micke P. Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade. Cancers. 2021; 13(13):3116. https://doi.org/10.3390/cancers13133116
Chicago/Turabian StyleEltahir, Mohamed, Johan Isaksson, Johanna Sofia Margareta Mattsson, Klas Kärre, Johan Botling, Martin Lord, Sara M. Mangsbo, and Patrick Micke. 2021. "Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade" Cancers 13, no. 13: 3116. https://doi.org/10.3390/cancers13133116
APA StyleEltahir, M., Isaksson, J., Mattsson, J. S. M., Kärre, K., Botling, J., Lord, M., Mangsbo, S. M., & Micke, P. (2021). Plasma Proteomic Analysis in Non-Small Cell Lung Cancer Patients Treated with PD-1/PD-L1 Blockade. Cancers, 13(13), 3116. https://doi.org/10.3390/cancers13133116