
A Systems Approach to Brain Tumor Treatment


 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
Highlights
- Brain tumors are difficult to treat because inter-patient and intratumoral heterogeneity are particularly pronounced. Moreover, the blood–brain barrier (BBB) presents an impediment to drug delivery.
- Technological advancements now allow robust profiling of genomic, epigenomic, transcriptomic, proteomic, and metabolomic changes in brain tumors.
- In parallel, efficient computational approaches to probe multi-modal data extensively and to infer regulatory mechanisms distinguishing tumor cell subpopulations have fostered novel intervention strategies (e.g., targeted- and immunotherapies) to treat brain tumors effectively.
- To address the current gaps preventing the translation of these advancements into clinical practice, a procedural framework that incorporates sensitive diagnostic and prognostic tests and computational methodologies must be developed to analyze clinical molecular profiles, enable relevant patient stratification, and identify appropriate treatments for N-of-1 therapy selection.
1. Introduction
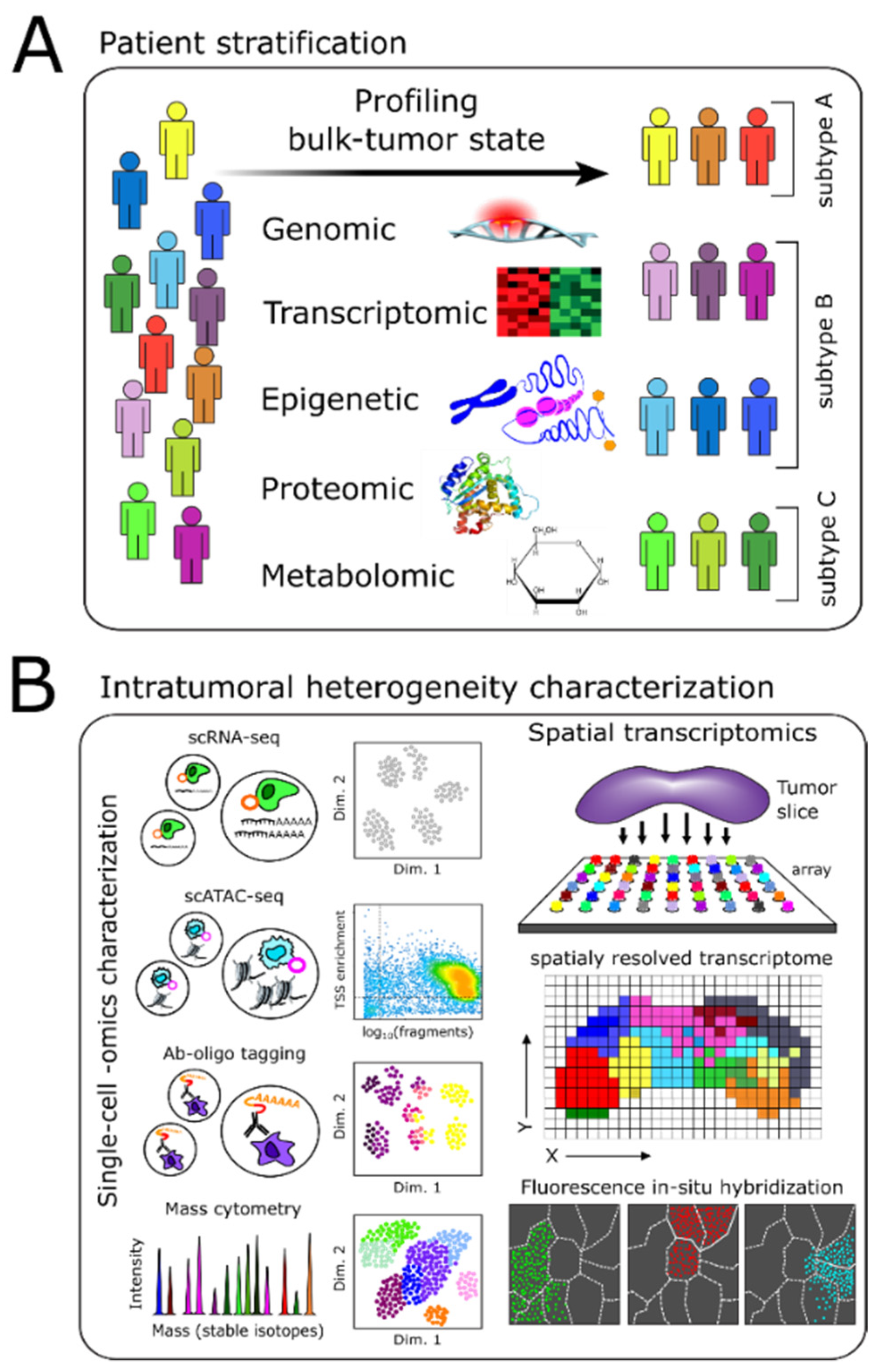
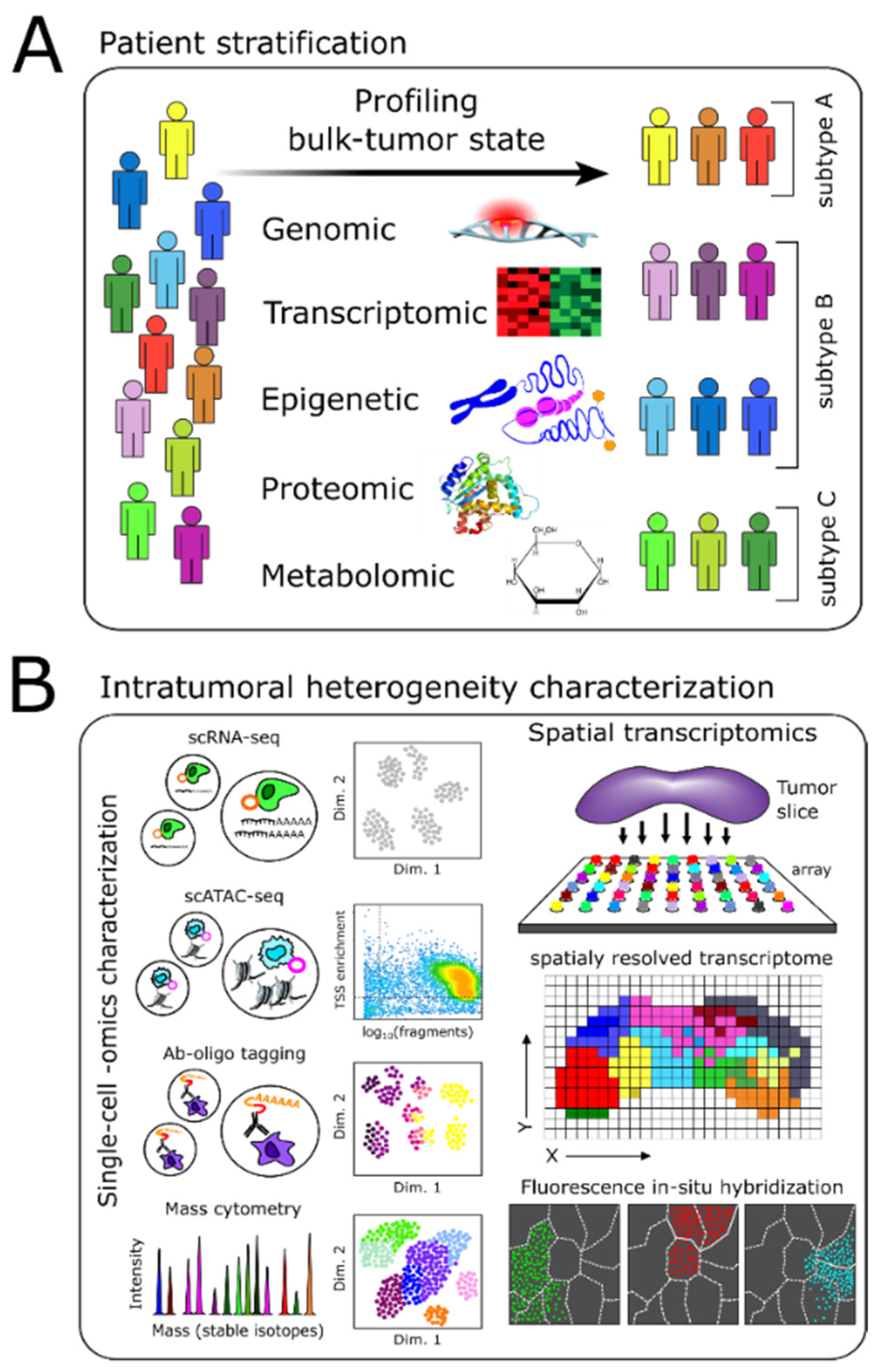
2. Molecular Profiling-Based Patient Stratification and Monitoring
2.1. Genome, Epigenome, and Transcriptome Characterization
2.2. Proteomics-Based Characterization
2.3. Metabolomics-Based Stratification and Monitoring
2.4. Liquid Biopsies and Longitudinal Monitoring of Patients
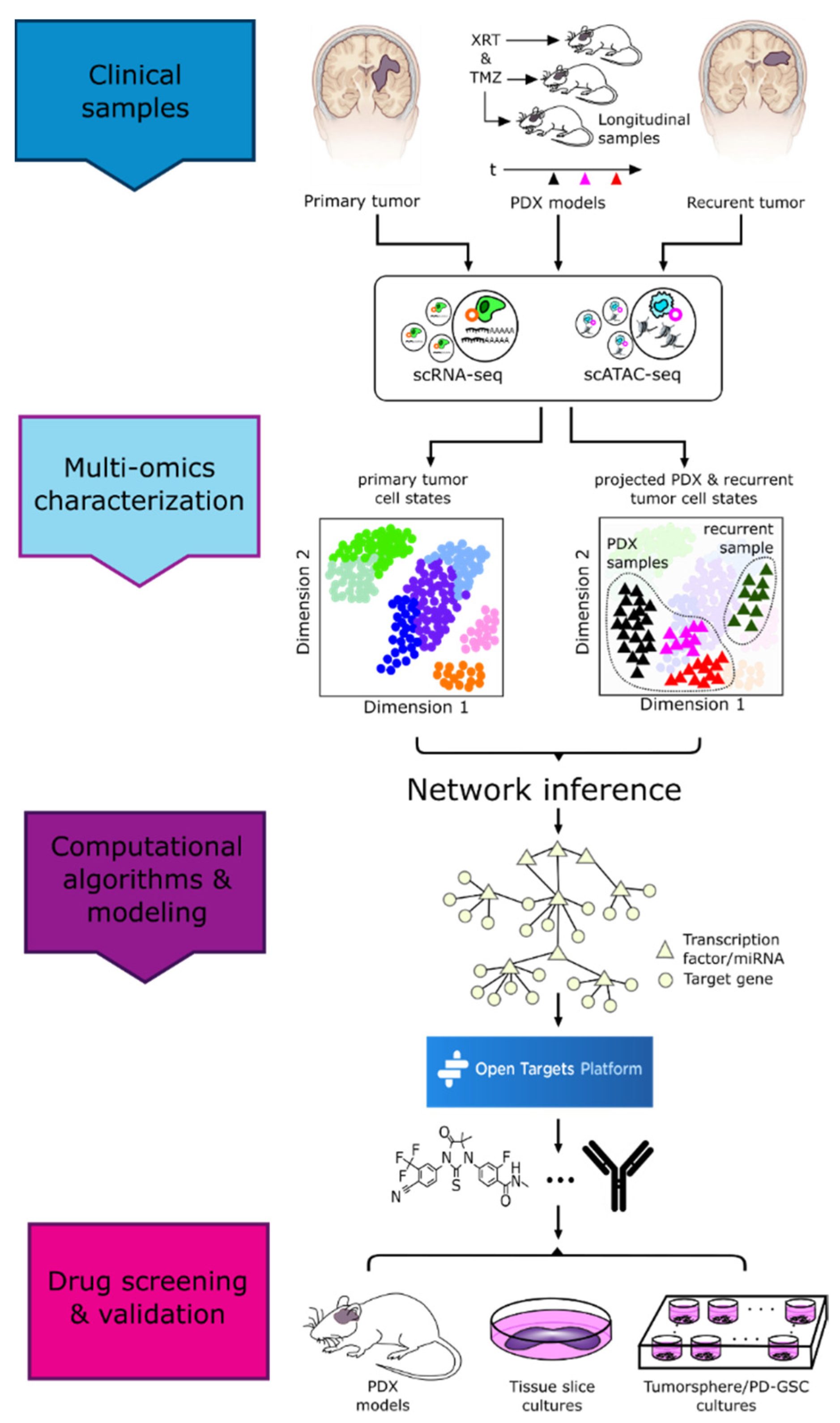
3. Intratumoral Heterogeneity
3.1. Methods Enabling Single-Cell Level Characterization of Tumors
3.2. Characterizing Genetic and Non-Genetic Cell–Cell Heterogeneity
3.2.1. Genetic Intratumoral Heterogeneity
3.2.2. Non-Genetic Intratumoral Heterogeneity
3.3. Spatial Heterogeneity
3.4. Tumor Microenvironment (TME)
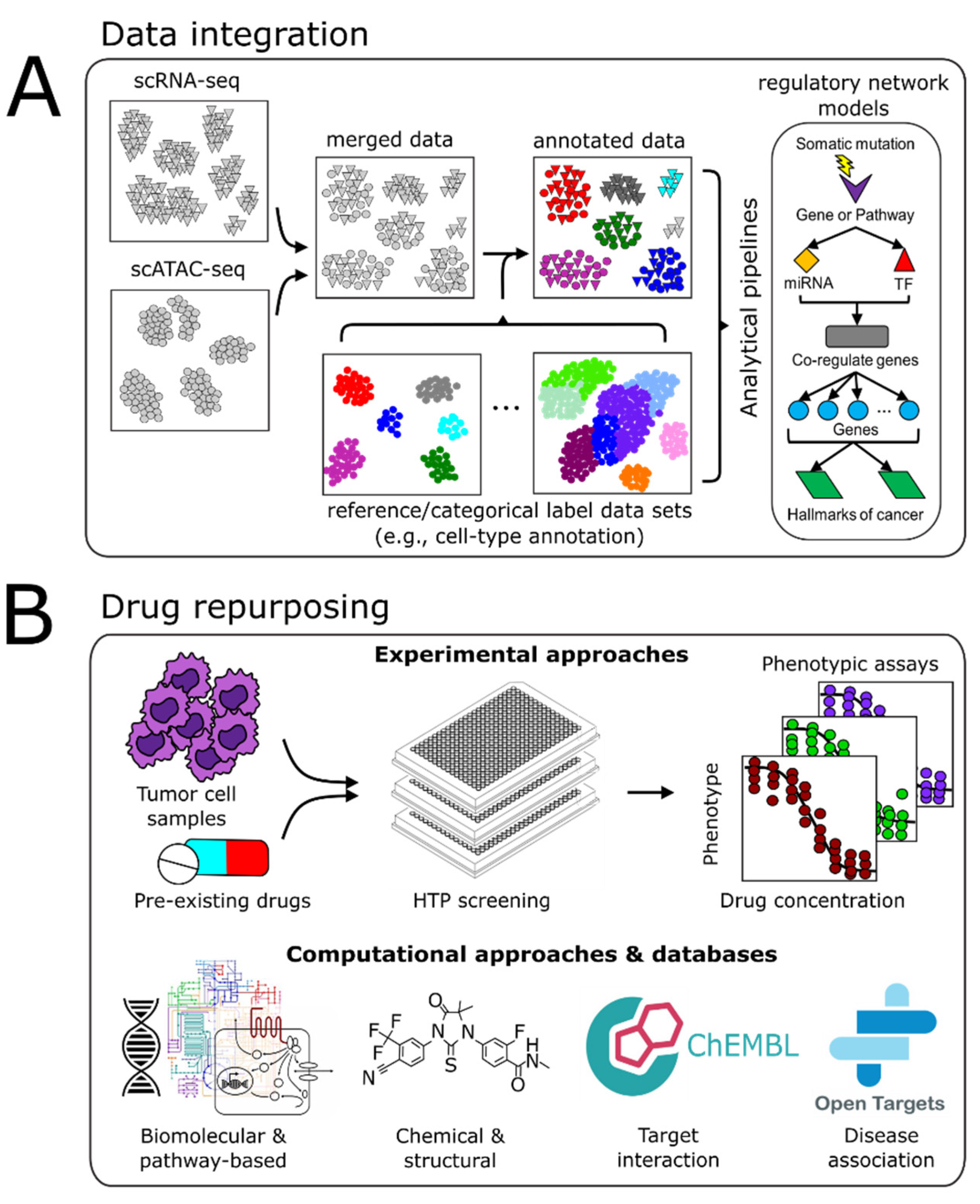
4. Vertical Data Integration and Computational Analyses
4.1. Batch Integration
4.2. Multimodal Data Integration
4.3. Computational Pipelines
5. Drug Repurposing
5.1. Experimental Approaches
5.2. Computational Approaches
5.3. Drug-Relevant Databases
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.; Magill, S.T.; Aghi, M.K. Molecularly targeted therapies for recurrent glioblastoma: Current and future targets. Neurosurg. Focus 2014, 37, E15. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarker. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma-are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Lippitz, B.; Lindquist, C.; Paddick, I.; Peterson, D.; O’Neill, K.; Beaney, R. Stereotactic radiosurgery in the treatment of brain metastases: The current evidence. Cancer Treat. Rev. 2014, 40, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Stockham, A.L.; Suh, J.H.; Chao, S.T.; Barnett, G.H. Management of recurrent brain metastasis after radiosurgery. Prog. Neurol. Surg. 2012, 25, 273–286. [Google Scholar] [CrossRef]
- Ajithkumar, T.; Parkinson, C.; Fife, K.; Corrie, P.; Jefferies, S. Evolving treatment options for melanoma brain metastases. Lancet Oncol. 2015, 16, e486–e497. [Google Scholar] [CrossRef]
- Fan, Q.-W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR Phosphorylates Tumor-Derived EGFRvIII Driving STAT3/5 and Progression in Glioblastoma. Cancer Cell 2013, 24, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Keunen, O.; Taxt, T.; Grüner, R.; Lund-Johansen, M.; Tonn, J.C.; Pavlin, T.; Bjerkvig, R.; Niclou, S.P.; Thorsen, F. Multimodal imaging of gliomas in the context of evolving cellular and molecular therapies. Adv. Drug Deliv. Rev. 2014, 76, 98–115. [Google Scholar] [CrossRef] [Green Version]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Somerville, R.P.T.; Shoshan, Y.; Eng, C.; Barnett, G.; Miller, D.; Cowell, J.K. Molecular analysis of two putative tumour suppressor genes, PTEN and DMBT, which have been implicated in glioblastoma multiforme disease progression. Oncogene 1998, 17, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, J.; Yang, J.; Pan, T.; Zhang, S.; Wang, Z. MiR-21 protected human glioblastoma U87MG cells from chemotherapeutic drug temozolomide induced apoptosis by decreasing Bax/Bcl-2 ratio and caspase-3 activity. Brain Res. 2010, 1352, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, D.; Mikkelsen, T.; Schwechheimer, K.; Rosenblum, M.L.; Cavanee, W.; Vogelstein, B. Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355, 846–847. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Barnett, G.H.; Hara, H.; Morimura, T.; Takeuchi, J. MDM2 protein confers the resistance of a human glioblastoma cell line to cisplatin-induced apoptosis. Oncogene 1995, 10, 2001–2006. [Google Scholar]
- Marte, B. Tumour heterogeneity. Nature 2013, 501, 327. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Hood, L. Personalized, Precision, and N-of-One Medicine: A Clarification of Terminology and Concepts. Perspect. Biol. Med. 2019, 62, 617–639. [Google Scholar] [CrossRef]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [Green Version]
- Haar, C.P.; Hebbar, P.; Wallace IV, G.C.; Das, A.; Vandergrift, W.A.; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug resistance in glioblastoma: A mini review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef]
- Richard, A.; Boullu, L.; Herbach, U.; Bonnafoux, A.; Morin, V.; Vallin, E.; Guillemin, A.; Papili Gao, N.; Gunawan, R.; Cosette, J.; et al. Single-Cell-Based Analysis Highlights a Surge in Cell-to-Cell Molecular Variability Preceding Irreversible Commitment in a Differentiation Process. PLoS Biol. 2016, 14, 1–35. [Google Scholar] [CrossRef]
- Mojtahedi, M.; Skupin, A.; Zhou, J.; Casta, I.G.; Rebecca, Y.; Chang, H.; Trachana, K.; Giuliani, A.; Huang, S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016, 14, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Levin, V.A.; Tonge, P.J.; Gallo, J.M.; Birtwistle, M.R.; Dar, A.C.; Iavarone, A.; Paddison, P.J.; Heffron, T.P.; Elmquist, W.F.; Lachowicz, J.E.; et al. CNS Anticancer Drug Discovery and Development Conference White Paper. Neuro Oncol. 2015, 17, vi1–vi26. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Angulo, A.M.; Hennessy, B.T.J.; Mills, G.B. Future of Personalized Medicine in Oncology: A Systems Biology Approach. J. Clin. Oncol. 2010, 28, 2777–2783. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Archer, T.C.; Fertig, E.J.; Gosline, S.J.C.; Hafner, M.; Hughes, S.K.; Joughin, B.A.; Meyer, A.S.; Piccolo, S.R.; Shajahan-Haq, A.N. Systems Approaches to Cancer Biology. Cancer Res. 2016, 76, 6774–6777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.S.; Alderete, B.; Minn, Y.; Borell, T.J.; Perry, A.; Mohapatra, G.; Hosek, S.M.; Kimmel, D.; O’Fallon, J.; Yates, A.; et al. Localization of common deletion regions on 1p and 19q in human gliomas and their association with histological subtype. Oncogene 1999, 18, 4144–4152. [Google Scholar] [CrossRef] [Green Version]
- Josefa Bello, M.; Leone, P.E.; Vaquero, J.; De Campos, J.M.; Elena Kusak, M.; Sarasa, J.L.; Pestaña, A.; Rey, J.A. Allelic loss at 1p and 19q frequently occurs in association and may represent early oncogenic events in oligodendroglial tumors. Int. J. Cancer 1995, 64, 207–210. [Google Scholar] [CrossRef]
- Sabha, N.; Knobbe, C.B.; Maganti, M.; Al Omar, S.; Bernstein, M.; Cairns, R.; Çako, B.; von Deimling, A.; Capper, D.; Mak, T.W.; et al. Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol. 2014, 16, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Lewandowska, M.A.; Furtak, J.; Szylberg, T.; Roszkowski, K.; Windorbska, W.; Rytlewska, J.; Jóźwicki, W. An Analysis of the Prognostic Value of IDH1 (Isocitrate Dehydrogenase 1) Mutation in Polish Glioma Patients. Mol. Diagn. Ther. 2014, 18, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Tachibana, I.; Passe, S.M.; Huntley, B.K.; Borell, T.J.; Iturria, N.; O’Fallon, J.R.; Schaefer, P.L.; Scheithauer, B.W.; James, C.D.; et al. PTEN Mutation, EGFR Amplification, and Outcome in Patients with Anaplastic Astrocytoma and Glioblastoma Multiforme. JNCI J. Natl. Cancer Inst. 2001, 93, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Gil-Benso, R.; Lopez-Gines, C.; Benito, R.; López-Guerrero, J.A.; Callaghan, R.C.; Pellín, A.; Roldán, P.; Cerdá-Nicolas, M. Concurrent EGFR amplification and TP-53 mutation in glioblastomas. Clin. Neuropathol. 2007, 26, 224–231. [Google Scholar] [CrossRef]
- Eckel-Passow, J.E.; Lachance, D.H.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Sicotte, H.; Pekmezci, M.; Rice, T.; Kosel, M.L.; Smirnov, I.V.; et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N. Engl. J. Med. 2015, 372, 2499–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Zlatescu, M.C.; Betensky, R.A.; Ino, Y.; Cairncross, J.G.; Louis, D.N. PTEN Is a Target of Chromosome 10q Loss in Anaplastic Oligodendrogliomas and PTEN Alterations Are Associated with Poor Prognosis. Am. J. Pathol. 2001, 159, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Tosoni, A.; Franceschi, E.; Poggi, R.; Brandes, A.A. Relapsed Glioblastoma: Treatment Strategies for Initial and Subsequent Recurrences. Curr. Treat. Options Oncol. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Marzese, D.M.; Witz, I.P.; Kelly, D.F.; Hoon, D.S.B. Epigenomic landscape of melanoma progression to brain metastasis: Unexplored therapeutic alternatives. Epigenomics 2015, 7, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Marzese, D.M.; Scolyer, R.A.; Roqué, M.; Vargas-Roig, L.M.; Huynh, J.L.; Wilmott, J.S.; Murali, R.; Buckland, M.E.; Barkhoudarian, G.; Thompson, J.F.; et al. DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro Oncol. 2014, 16, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469. [Google Scholar] [CrossRef]
- Orozco, J.I.J.; Knijnenburg, T.A.; Manughian-Peter, A.O.; Salomon, M.P.; Barkhoudarian, G.; Jalas, J.R.; Wilmott, J.S.; Hothi, P.; Wang, X.; Takasumi, Y.; et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat. Commun. 2018, 9, 4627. [Google Scholar] [CrossRef]
- Guilhamon, P.; Kushida, M.M.; Macleod, G.; Am, S.; Tabori, U.; Taylor, M.D.; Haibe-kains, B.; Angers, S. Chromatin Blueprint of Glioblastoma Stem Cells Reveals. bioRxiv 2018. [Google Scholar] [CrossRef]
- Guilhamon, P.; Chesnelong, C.; Kushida, M.M.; Nikolic, A.; Singhal, D.; Macleod, G.; Tonekaboni, S.A.M.; Cavalli, F.M.G.; Arlidge, C.; Rajakulendran, N.; et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. Elife 2021, 10, 1–20. [Google Scholar] [CrossRef]
- Cayer, D.M.; Nazor, K.L.; Schork, N.J. Mission critical: The need for proteomics in the era of next-generation sequencing and precision medicine. Hum. Mol. Genet. 2016, 25, R182–R189. [Google Scholar] [CrossRef]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes with Different Clinical Outcome. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Sakaida, T.; Hiwasa, T.; Nagai, Y.; Ishikura, H.; Takiguchi, M.; Yamaura, A. Molecular Classification and Survival Prediction in Human Gliomas Based on Proteome Analysis. Cancer Res. 2004, 64, 2496–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinina, J.; Peng, J.; Ritchie, J.C.; Van Meir, E.G. Proteomics of gliomas: Initial biomarker discovery and evolution of technology. Neuro Oncol. 2011, 13, 926–942. [Google Scholar] [CrossRef] [PubMed]
- Hristova, V.A.; Chan, D.W. Cancer biomarker discovery and translation: Proteomics and beyond. Expert Rev. Proteomics 2019, 16, 93–103. [Google Scholar] [CrossRef]
- Arora, A.; Patil, V.; Kundu, P.; Kondaiah, P.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Pal, D.; Somasundaram, K. Serum biomarkers identification by iTRAQ and verification by MRM: S100A8/S100A9 levels predict tumor-stroma involvement and prognosis in Glioblastoma. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 2012, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Miyauchi, E.; Furuta, T.; Ohtsuki, S.; Tachikawa, M.; Uchida, Y.; Sabit, H.; Obuchi, W.; Baba, T.; Watanabe, M.; Terasaki, T.; et al. Identification of blood biomarkers in glioblastoma by SWATH mass spectrometry and quantitative targeted absolute proteomics. PLoS ONE 2018, 13, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeure, K.; Fack, F.; Duriez, E.; Tiemann, K.; Bernard, A.; Golebiewska, A.; Bougnaud, S.; Bjerkvig, R.; Domon, B.; Niclou, S.P. Targeted proteomics to assess the response to anti-Angiogenic treatment in human Glioblastoma (GBM). Mol. Cell. Proteom. 2016, 15, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Shahi, P.; Kim, S.C.; Haliburton, J.R.; Gartner, Z.J.; Abate, A.R. Abseq: Ultrahigh-throughput single cell protein profiling with droplet microfluidic barcoding. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Cheng, A.; Montalbano, A.; Hao, S.; Stoeckius, M.; Legut, M.; Roush, T.; Herrera, A.; Papalexi, E.; Ouyang, Z.; et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 2019, 16, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salzillo, T.C.; Hu, J.; Nguyen, L.; Whiting, N.; Lee, J.; Weygand, J.; Dutta, P.; Pudakalakatti, S.; Millward, N.Z.; Gammon, S.T.; et al. Interrogating Metabolism in Brain Cancer. Magn. Reson. Imaging Clin. N. Am. 2016, 24, 687–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.M.; Parolia, A.; Dunphy, M.P.; Venneti, S. Non-invasive metabolic imaging of brain tumours in the era of precision medicine. Nat. Rev. Clin. Oncol. 2016, 13, 725–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Deberardinis, R.J. Analyzing Tumor Metabolism In Vivo. Ann. Rev. Cancer Biol. 2016, 1, 99–117. [Google Scholar] [CrossRef]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2019, 20, 57–70. [Google Scholar] [CrossRef]
- Antoniewicz, M.R. A guide to 13C metabolic flux analysis for the cancer biologist. Exp. Mol. Med. 2018, 50, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426. [Google Scholar] [CrossRef]
- Pérez-Callejo, D.; Romero, A.; Provencio, M.; Torrente, M. Liquid biopsy based biomarkers in non-small cell lung cancer for diagnosis and treatment monitoring. Transl. Lung Cancer Res. 2016, 5, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Saxena, D.; Sheikh, S.; Kao, G.; Binder, Z.A.; Alonso-Basanta, M.; O’Rourke, D.M.; Nasrallah, M.P.; Dorsey, J.F. Rapid and ultrasensitive digital PCR (dPCR) profiling of EGFRvIII in tumor cells and tissues. Neuro-Oncol. Adv. 2019, 1. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating Cancer of the Central Nervous System Through Next-Generation Sequencing of Cerebrospinal Fluid. J. Clin. Oncol. 2016, 34, 2404–2415. [Google Scholar] [CrossRef]
- Boire, A.; Brandsma, D.; Brastianos, P.K.; Le Rhun, E.; Ahluwalia, M.; Junck, L.; Glantz, M.; Groves, M.D.; Lee, E.Q.; Lin, N.; et al. Liquid biopsy in central nervous system metastases: A RANO review and proposals for clinical applications. Neuro Oncol. 2019, 21, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Merker, J.D.; Oxnard, G.R.; Compton, C.; Diehn, M.; Hurley, P.; Lazar, A.J.; Lindeman, N.; Lockwood, C.M.; Rai, A.J.; Schilsky, R.L.; et al. Circulating Tumor DNA Analysis in Patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. Arch. Pathol. Lab. Med. 2018, 142, 1242–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, S.; Nabavizadeh, S.A.; Mays, J.; Till, J.E.; Ware, J.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.J.; Guiry, S.; et al. Clinical utility of plasma cell-free DNA in adult patients with newly diagnosed glioblastoma—A pilot prospective study. Clin. Cancer Res. 2019, 26, 397–407. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Wang, Y.; Navin, N.E. Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 2015, 58, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-cell RNA-seq technologies and related computational data analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakken, T.E.; Hodge, R.D.; Miller, J.A.; Yao, Z.; Nguyen, T.N.; Aevermann, B.; Barkan, E.; Bertagnolli, D.; Casper, T.; Dee, N.; et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 2018, 13, e0209648. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Slyper, M.; Porter, C.B.M.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef]
- Thrupp, N.; Sala Frigerio, C.; Wolfs, L.; Skene, N.G.; Fattorelli, N.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; Mancuso, R.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016, 17, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Levsky, J.M.; Singer, R.H. Fluorescence in situ hybridization: Past, present and future. J. Cell Sci. 2003, 116, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 1467, 1463–1467. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Deng, Y.; Su, G.; Enninful, A.; Guo, C.C.; Tebaldi, T.; Zhang, D.; Kim, D.; Bai, Z.; et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 2020, 183, 1665–1681.e18. [Google Scholar] [CrossRef]
- Brock, A.; Chang, H.; Huang, S. Non-genetic heterogeneity—A mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet. 2009, 10, 336. [Google Scholar] [CrossRef]
- Huang, S. Genetic and non-genetic instability in tumor progression: Link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013, 32, 423–448. [Google Scholar] [CrossRef]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Reimand, J.; Lan, X.; Head, R.; Zhu, X.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The Evidence of Glioblastoma Heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor Tyrosine Kinase Genes Amplified in Glioblastoma Exhibit a Mutual Exclusivity in Variable Proportions Reflective of Individual Tumor Heterogeneity. Cancer Res. 2012, 72, 1614–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Huang, S. Reconciling Non-Genetic Plasticity with Somatic Evolution in Cancer. Trends Cancer 2021, 7, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Valind, A.; Holmquist Mengelbier, L.; Bredin, S.; Cornmark, L.; Jansson, C.; Wali, A.; Staaf, J.; Viklund, B.; Øra, I.; et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat. Genet. 2018, 50, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Sun, R.; Curtis, C. A population genetics perspective on the determinants of intra-tumor heterogeneity. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 109–126. [Google Scholar] [CrossRef] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, S.G.M.; Combi, R.; Cajola, L.; Patrizi, A.; Redaelli, S.; Bentivegna, A.; Baronchelli, S.; Maira, G.; Pollo, B.; Mangiola, A.; et al. Distinct pools of cancer stem-like cells coexist within human glioblastomas and display different tumorigenicity and independent genomic evolution. Oncogene 2009, 28, 1807–1811. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, S.; Buart, S.; Chouaib, S. Hypoxic Stress-Induced Tumor and Immune Plasticity, Suppression, and Impact on Tumor Heterogeneity. Front. Immunol. 2017, 8, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, T.M.; Carrillo, J.A.; Achrol, A.A.; Salomon, M.P.; Marzese, D.M.; Park, J.H.; Baliga, N.S.; Kesari, S. Understanding the brain tumor microenvironment: Considerations to applying systems biology and immunotherapy. Int. J. Neurooncol. 2018, 1, 25–33. [Google Scholar] [CrossRef]
- Zhou, J.X.; Taramelli, R.; Pedrini, E.; Knijnenburg, T.; Huang, S. Extracting Intercellular Signaling Network of Cancer Tissues using Ligand-Receptor Expression Patterns from Whole-tumor and Single-cell Transcriptomes. Sci. Rep. 2017, 7, 8815. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, H.; Roncali, L.; Rousseau, A.; Chérel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 1–21. [Google Scholar] [CrossRef]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Kennedy, B.C.; Showers, C.R.; Anderson, D.E.; Anderson, L.; Canoll, P.; Bruce, J.N.; Anderson, R.C.E. Tumor-associated macrophages in glioma: Friend or foe? J. Oncol. 2013, 2013, 486912. [Google Scholar] [CrossRef]
- Leblond, M.M.; Pérès, E.A.; Helaine, C.; Gérault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget 2017, 8, 72597–72612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Luo, F.; Yang, J.; Liu, J.; Liu, R.; Wang, L.; Wang, C.; Deng, Y.; Lu, Z.; Wang, Y.; et al. TLR2 promotes glioma immune evasion by downregulating MHC class II molecules in microglia. Cancer Immunol. Res. 2018, 6, 1220–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T. svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014, 42, e161. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef]
- Forcato, M.; Romano, O.; Bicciato, S. Computational methods for the integrative analysis of single-cell data. Brief. Bioinform. 2020, 22, 20–29. [Google Scholar] [CrossRef]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [Green Version]
- Hie, B.; Bryson, B.; Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019, 37, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wu, Y.; Tian, W. A novel approach to remove the batch effect of single-cell data. Cell Discov. 2019, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Barkas, N.; Petukhov, V.; Nikolaeva, D.; Lozinsky, Y.; Demharter, S.; Khodosevich, K.; Kharchenko, P.V. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods 2019, 16, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Polański, K.; Young, M.D.; Miao, Z.; Meyer, K.B.; Teichmann, S.A.; Park, J.-E. BBKNN: Fast batch alignment of single cell transcriptomes. Bioinformatics 2019, 36, 964–965. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hoinka, J.; Przytycka, T.M. Subpopulation Detection and Their Comparative Analysis across Single-Cell Experiments with scPopCorn. Cell Syst. 2019, 8, 506–513.e5. [Google Scholar] [CrossRef] [Green Version]
- Leonavicius, K.; Nainys, J.; Kuciauskas, D.; Mazutis, L. Multi-omics at single-cell resolution: Comparison of experimental and data fusion approaches. Curr. Opin. Biotechnol. 2019, 55, 159–166. [Google Scholar] [CrossRef]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR: An integrative and scalable software package for single-cell chromatin accessibility analysis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dekker, L.J.M.; Kannegieter, N.M.; Haerkens, F.; Toth, E.; Kros, J.M.; Steenhoff Hov, D.A.; Fillebeen, J.; Verschuren, L.; Leenstra, S.; Ressa, A.; et al. Multiomics profiling of paired primary and recurrent glioblastoma patient tissues. Neuro-Oncol. Adv. 2020, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef] [Green Version]
- Plaisier, C.L.; O’Brien, S.; Bernard, B.; Reynolds, S.; Simon, Z.; Toledo, C.M.; Ding, Y.; Reiss, D.J.; Paddison, P.J.; Baliga, N.S. Causal Mechanistic Regulatory Network for Glioblastoma Deciphered Using Systems Genetics Network Analysis. Cell Syst. 2016, 3, 172–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: Computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- Karchin, R.; Diekhans, M.; Kelly, L.; Thomas, D.J.; Pieper, U.; Eswar, N.; Haussler, D.; Sali, A. LS-SNP: Large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics 2005, 21, 2814–2820. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Krishnan, V.G.; Mort, M.E.; Xin, F.; Kamati, K.K.; Cooper, D.N.; Mooney, S.D.; Radivojac, P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 2009, 25, 2744–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. U.S. Food and Drug Adminstation Fact Sheet. Available online: https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance (accessed on 21 June 2021).
- National Cancer Institute National Cancer Institute Cancer Treatments. Available online: https://www.cancer.gov/about-cancer/treatment/drugs (accessed on 21 June 2021).
- Mullard, A. 2020 FDA drug approvals. Nat. Rev. Drug Discov. 2021, 20, 85–90. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Iljin, K.; Ketola, K.; Vainio, P.; Halonen, P.; Kohonen, P.; Fey, V.; Grafström, R.C.; Perälä, M.; Kallioniemi, O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6070–6078. [Google Scholar] [CrossRef] [Green Version]
- Hothi, P.; Martins, T.J.; Chen, L.; Deleyrolle, L.; Yoon, J.-G.; Reynolds, B.; Foltz, G. High-Throughput Chemical Screens Identify Disulfiram as an Inhibitor of Human Glioblastoma Stem Cells. Oncotarget 2012, 3, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salentin, S.; Adasme, M.F.; Heinrich, J.C.; Haupt, V.J.; Daminelli, S.; Zhang, Y.; Schroeder, M. From malaria to cancer: Computational drug repositioning of amodiaquine using PLIP interaction patterns. Sci. Rep. 2017, 7, 11401. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; He, D.; Qiu, Y.; Krawczuk, P.; Sun, X.; Xie, L. Rational discovery of dual-indication multi-target PDE/Kinase inhibitor for precision anti-cancer therapy using structural systems pharmacology. PLOS Comput. Biol. 2019, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tanoli, Z.; Seemab, U.; Scherer, A.; Wennerberg, K.; Tang, J.; Vähä-Koskela, M. Exploration of databases and methods supporting drug repurposing: A comprehensive survey. Brief. Bioinform. 2020, 22, 1656–1678. [Google Scholar] [CrossRef] [Green Version]
- Zagidullin, B.; Aldahdooh, J.; Zheng, S.; Wang, W.; Wang, Y.; Saad, J.; Malyutina, A.; Jafari, M.; Tanoli, Z.; Pessia, A.; et al. DrugComb: An integrative cancer drug combination data portal. Nucleic Acids Res. 2019, 47, W43–W51. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: Mining clinically relevant drug–gene interactions. Nucleic Acids Res. 2015, 44, D1036–D1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug–target identification and prioritisation. Nucleic Acids Res. 2020, 49, D1302–D1310. [Google Scholar] [CrossRef]
- Yang, H.; Qin, C.; Li, Y.H.; Tao, L.; Zhou, J.; Yu, C.Y.; Xu, F.; Chen, Z.; Zhu, F.; Chen, Y.Z. Therapeutic target database update 2016: Enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2015, 44, D1069–D1074. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Askoxylakis, V.; Guo, Y.; Datta, M.; Kloepper, J.; Ferraro, G.B.; Bernabeu, M.O.; Fukumura, D.; McDannold, N.; Jain, R.K. Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood–tumor barrier disruption. Proc. Natl. Acad. Sci. USA 2018, 115, E8717–E8726. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Park, J.H.; Feroze, A.H.; Emerson, S.N.; Mihalas, A.B.; Keene, C.D.; Cimino, P.J.; de Lomana, A.L.G.; Kannan, K.; Wu, W.-J.; Turkarslan, S.; et al. A single-cell based precision medicine approach using glioblastoma patient-specific models. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wouters, R.; Bevers, S.; Riva, M.; De Smet, F.; Coosemans, A. Immunocompetent mouse models in the search for effective immunotherapy in glioblastoma. Cancers 2021, 13, 19. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.J.; Furlan, S.N.; Mihalas, A.B.; Kaya-Okur, H.S.; Feroze, A.H.; Emerson, S.N.; Zheng, Y.; Carson, K.; Cimino, P.J.; Keene, C.D.; et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat. Biotechnol. 2021, 1–6. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.H.; de Lomana, A.L.G.; Marzese, D.M.; Juarez, T.; Feroze, A.; Hothi, P.; Cobbs, C.; Patel, A.P.; Kesari, S.; Huang, S.; et al. A Systems Approach to Brain Tumor Treatment. Cancers 2021, 13, 3152. https://doi.org/10.3390/cancers13133152
Park JH, de Lomana ALG, Marzese DM, Juarez T, Feroze A, Hothi P, Cobbs C, Patel AP, Kesari S, Huang S, et al. A Systems Approach to Brain Tumor Treatment. Cancers. 2021; 13(13):3152. https://doi.org/10.3390/cancers13133152
Chicago/Turabian StylePark, James H., Adrian Lopez Garcia de Lomana, Diego M. Marzese, Tiffany Juarez, Abdullah Feroze, Parvinder Hothi, Charles Cobbs, Anoop P. Patel, Santosh Kesari, Sui Huang, and et al. 2021. "A Systems Approach to Brain Tumor Treatment" Cancers 13, no. 13: 3152. https://doi.org/10.3390/cancers13133152
APA StylePark, J. H., de Lomana, A. L. G., Marzese, D. M., Juarez, T., Feroze, A., Hothi, P., Cobbs, C., Patel, A. P., Kesari, S., Huang, S., & Baliga, N. S. (2021). A Systems Approach to Brain Tumor Treatment. Cancers, 13(13), 3152. https://doi.org/10.3390/cancers13133152