
NPM1 Mutational Status Underlines Different Biological Features in Pediatric AML

 , ,
, , 
Abstract
:Simple Summary
Abstract
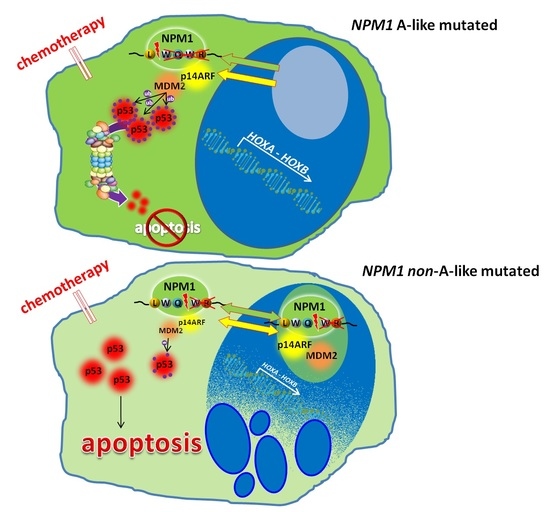
1. Introduction
2. Materials and Methods
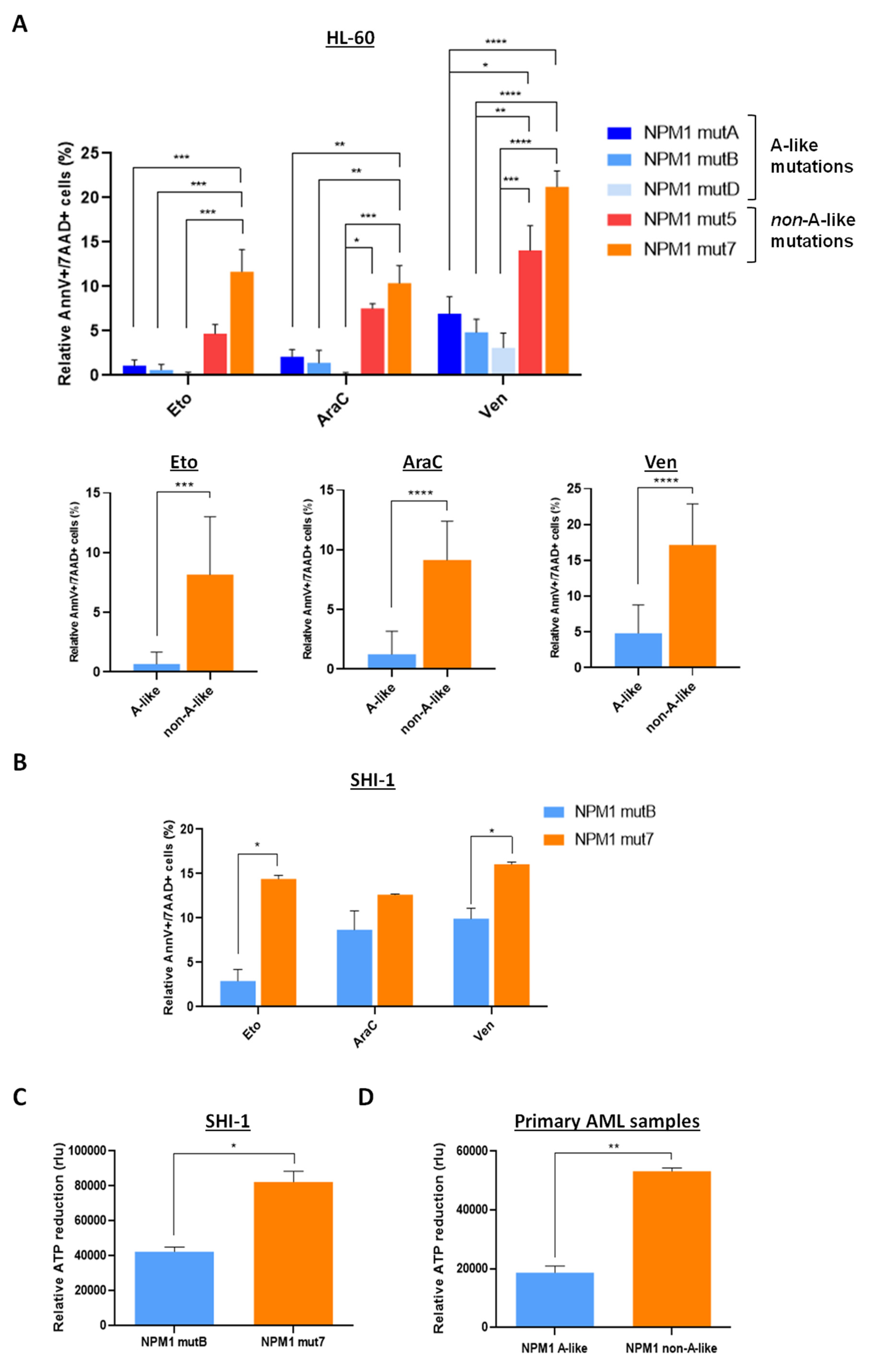
3. Results
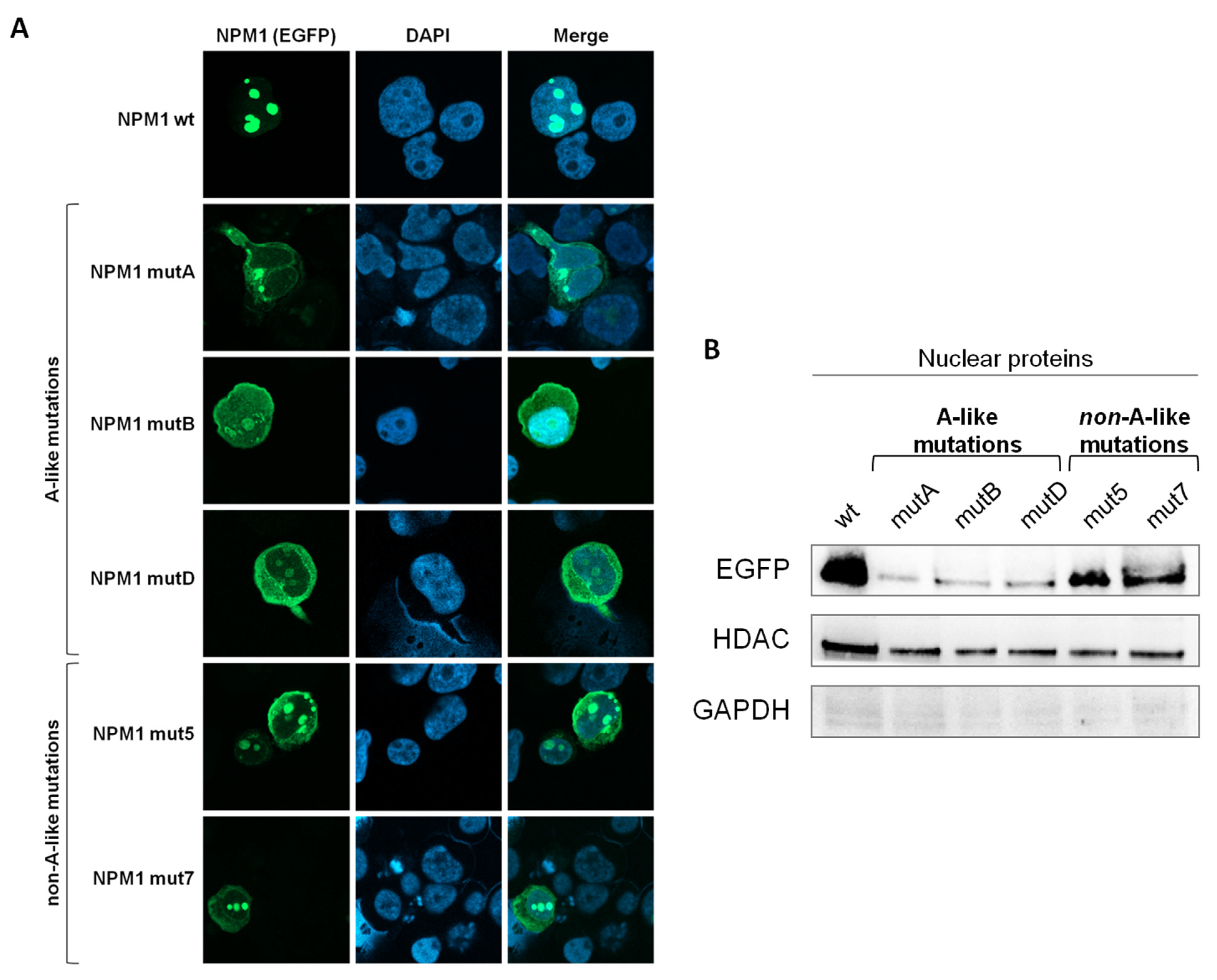
3.1. Intracellular Localization of the EGFP-NPM1 Mutants
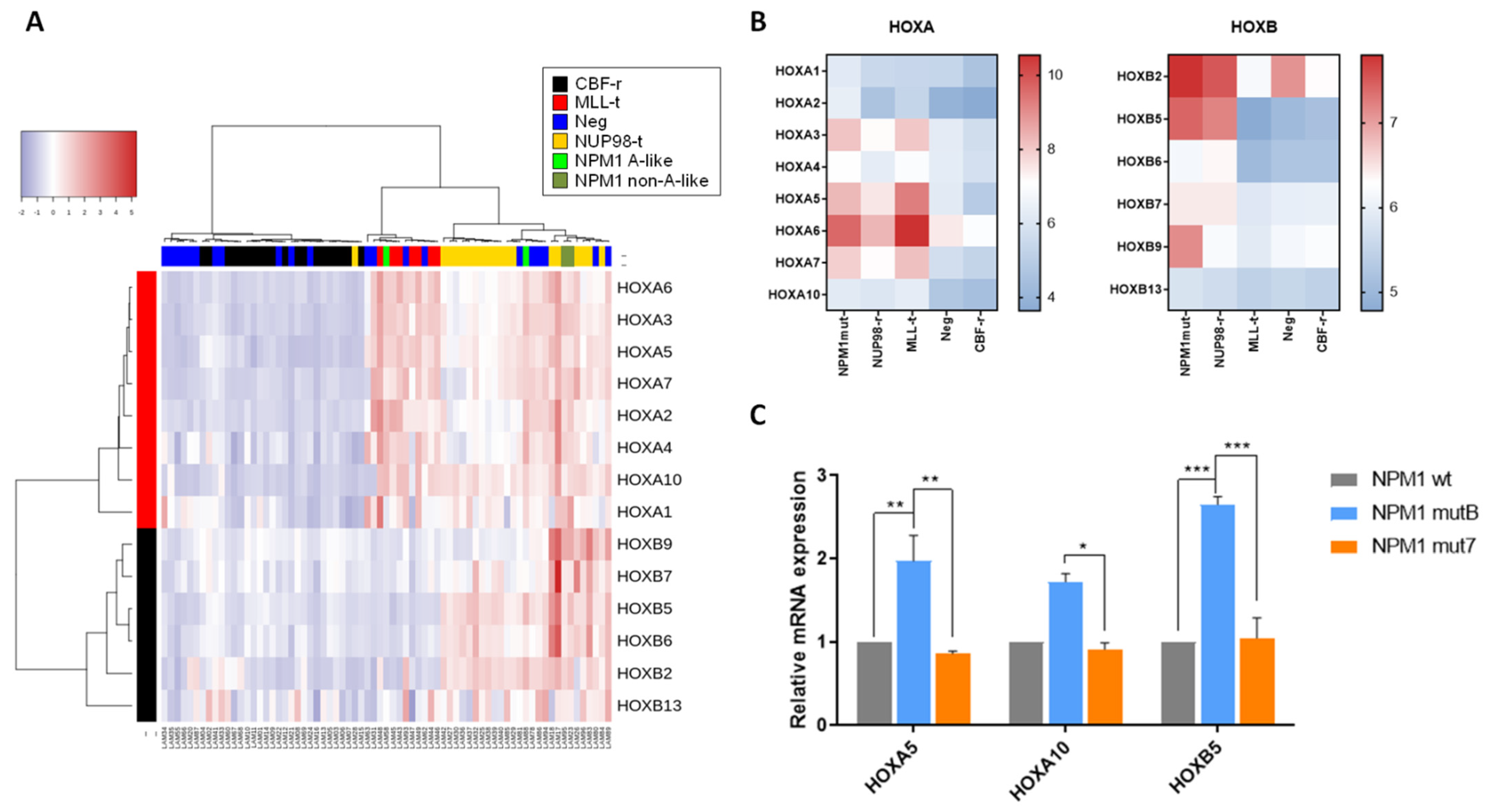
3.2. Effects of Different NPM1 Mutations on HOX Genes Expression
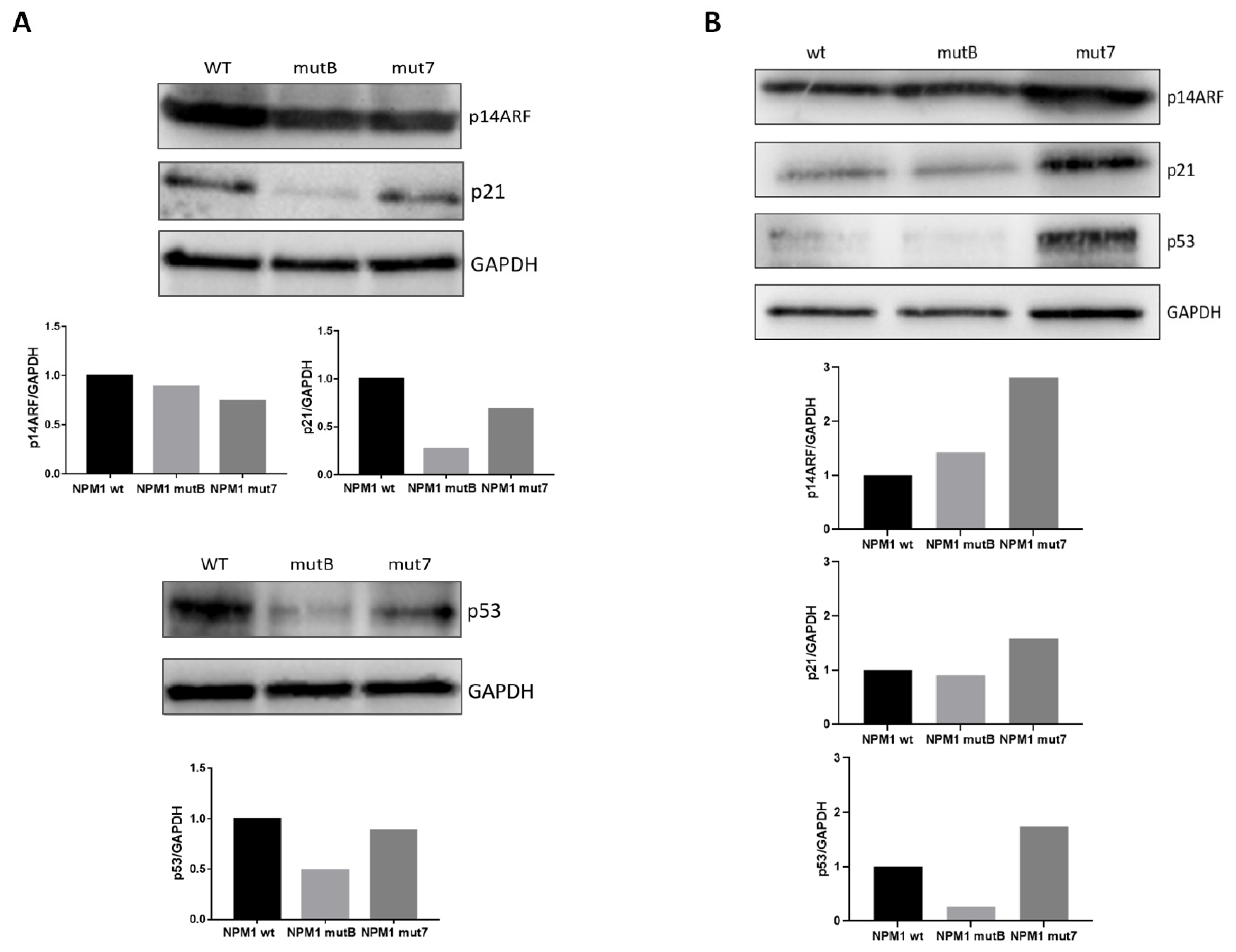
3.3. NPM1 Mutational Status and p53
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cordell, J.L.; Pulford, K.A.F.; Bigerna, B.; Roncador, G.; Banham, A.; Colombo, E.; Pelicci, P.G.; Mason, D.Y.; Falini, B. Detection of normal and chimeric nucleophosmin in human cells. Blood 1999, 93, 632–642. [Google Scholar] [CrossRef]
- Lam, Y.W.; Trinkle-Mulcahy, L.; Lamond, A.I. The nucleolus. J. Cell Sci. 2005, 118, 1335–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, M.; Lirussi, L.; Wilson, D.M.; Tell, G. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol. Biol. Cell 2014, 25, 1641–1652. [Google Scholar] [CrossRef]
- Vascotto, C.; Lirussi, L.; Poletto, M.; Tiribelli, M.; Damiani, D.; Fabbro, D.; Damante, G.; Demple, B.; Colombo, E.; Tell, G. Functional regulation of the apurinic/apyrimidinic endonuclease 1 by nucleophosmin: Impact on tumor biology. Oncogene 2014, 33, 2876–2887. [Google Scholar] [CrossRef] [PubMed]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, S.M.; Tan, B.X.; Ahmad, B.B.; Yan, T.; Chee, L.Y.; Ang, S.T.; Tay, K.G.; Koh, L.P.; Yeoh, A.E.J.; Koay, E.S.C.; et al. Mutant nucleophosmin deregulates cell death and myeloid differentiation through excessive caspase-6 and -8 inhibition. Blood 2010, 116, 3286–3296. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Bolli, N.; Liso, A.; Martelli, M.P.; Mannucci, R.; Pileri, S.; Nicoletti, I. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: Molecular basis and clinical implications. Leukemia 2009, 23, 1731–1743. [Google Scholar] [CrossRef]
- Arregi, I.; Falces, J.; Olazabal-Herrero, A.; Alonso-Mariño, M.; Taneva, S.G.; Rodríguez, J.A.; Urbaneja, M.A.; Bañuelos, S. Leukemia-associated mutations in nucleophosmin alter recognition by CRM1: Molecular basis of aberrant transport. PLoS ONE 2015, 10, e0130610. [Google Scholar] [CrossRef] [Green Version]
- Grummitt, C.G.; Townsley, F.M.; Johnson, C.M.; Warren, A.J.; Bycroft, M. Structural consequences of nucleophosmin mutations in acute myeloid leukemia. J. Biol. Chem. 2008, 283, 23326–23332. [Google Scholar] [CrossRef] [Green Version]
- Federici, L.; Falini, B. Nucleophosmin mutations in acute myeloid leukemia: A tale of protein unfolding and mislocalization. Protein Sci. 2013, 22, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Brown, P.; McIntyre, E.; Rau, R.; Meshinchi, S.; Lacayo, N.; Dahl, G.; Alonzo, T.A.; Chang, M.; Arceci, R.J.; Small, D. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood 2007, 110, 979–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollink, I.H.I.M.; Zwaan, C.M.; Zimmermann, M.; Arentsen-Peters, T.C.J.M.; Pieters, R.; Cloos, J.; Kaspers, G.J.L.; de Graaf, S.S.N.; Harbott, J.; Creutzig, U.; et al. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia 2009, 23, 262–270. [Google Scholar] [CrossRef]
- Pession, A.; Masetti, R.; Rizzari, C.; Putti, M.C.; Casale, F.; Fagioli, F.; Luciani, M.; Lo Nigro, L.; Menna, G.; Micalizzi, C.; et al. Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood 2013, 122, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzaniga, G.; Dell’Oro, M.G.; Mecucci, C.; Giarin, E.; Masetti, R.; Rossi, V.; Locatelli, F.; Martelli, M.F.; Basso, G.; Pession, A.; et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood 2005, 106, 1419–1422. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): Biologic and clinical features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.H.; Fang, J.P.; Liu, Y.C.; Jones, A.I.; Chai, L. Nucleophosmin mutations confer an independent favorable prognostic impact in 869 pediatric patients with acute myeloid leukemia. Blood Cancer J. 2020, 10, 1. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Kennedy, A.; Zhou, X.; Radtke, I.; Phillips, L.A.; Shurtleff, S.A.; Downing, J.R. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia 2007, 21, 2000–2009. [Google Scholar] [CrossRef]
- Alpermann, T.; Schnittger, S.; Eder, C.; Dicker, F.; Meggendorfer, M.; Kern, W.; Schmid, C.; Aul, C.; Staib, P.; Wendtner, C.M.; et al. Molecular subtypes of npm1 mutations have different clinical profiles, specific patterns of accompanying molecular mutations and varying outcomes in intermediate risk acute myeloid leukemia. Haematologica 2016, 101, e55–e58. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ohkubo, T.; Furuichi, Y.; Umekawa, H. Tryptophans 286 and 288 in the C-terminal Region of Protein B23.1 are Important for Its Nucleolar Localization. Biosci. Biotechnol. Biochem. 2002, 66, 2239–2242. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Bolli, N.; Shan, J.; Martelli, M.P.; Liso, A.; Pucciarini, A.; Bigerna, B.; Pasqualucci, L.; Mannucci, R.; Rosati, R.; et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood 2006, 107, 4514–4523. [Google Scholar] [CrossRef] [PubMed]
- Rau, R.; Brown, P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: Towards definition of a new leukaemia entity. Hematol. Oncol. 2009, 27, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, Y.; Park, J.; Bae, E.K.; Ahn, K.S.; Kim, I.; Bang, S.M.; Lee, J.H.; Yoon, S.S.; Lee, D.S.; Lee, Y.Y.; et al. Non-A type nucleophosmin 1 gene mutation predicts poor clinical outcome in de novo adult acute myeloid leukemia: Differential clinical importance of NPM1 mutation according to subtype. Int. J. Hematol. 2009, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Pastore, F.; Greif, P.A.; Schneider, S.; Ksienzyk, B.; Mellert, G.; Zellmeier, E.; Braess, J.; Sauerland, C.M.; Heinecke, A.; Krug, U.; et al. The NPM1 mutation type has no impact on survival in cytogenetically normal AML. PLoS ONE 2014, 9, e109759. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Bisio, V.; Zampini, M.; Tregnago, C.; Manara, E.; Salsi, V.; Di Meglio a Masetti, R.; Togni, M.; Di Giacomo, D.; Minuzzo, S.; Leszl a Zappavigna, V.; et al. NUP98-fusion transcripts characterize different biological entities within acute myeloid leukemia: A report from the AIEOP-AML group [Internet]. Leukemia 2017, 31, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Tregnago, C.; Manara, E.; Zampini, M.; Bisio, V.; Borga, C.; Bresolin, S.; Aveic, S.; Germano, G.; Basso, G.; Pigazzi, M. CREB engages C/EBPδ to initiate leukemogenesis. Leukemia 2016, 30, 1887–1896. [Google Scholar] [CrossRef]
- Nagy, Á.; Ősz, Á.; Budczies, J.; Krizsán, S.; Szombath, G.; Demeter, J.; Bödör, C.; Győrffy, B. Elevated HOX gene expression in acute myeloid leukemia is associated with NPM1 mutations and poor survival. J. Adv. Res. 2019, 20, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Hoster, E.; Unterhalt, M.; Schneider, S.; Dufour, A.; Benthaus, T.; Mellert, G.; Zellmeier, E.; Bohlander, S.K.; Feuring-Buske, M.; et al. NPM1 but not FLT3-ITD mutations predict early blast cell clearance and CR rate in patients with normal karyotype AML (NK-AML) or high-risk myelodysplastic syndrome (MDS). Blood 2009, 113, 5250–5253. [Google Scholar] [CrossRef] [Green Version]
- Borrow, J.; Dyer, S.A.; Akiki, S.; Griffiths, M.J. Molecular roulette: Nucleophosmin mutations in AML are orchestrated through N-nucleotide addition by TdT. Blood 2019, 134, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Selim, D.; Alonzo, T.A.; Othus, M.; Gerbing, R.; Ostronoff, F.; Tarlock, K.; Kutny, M.A.; Aplenc, R.; Kolb, E.A.; Radich, J.P.; et al. Genomic Subtypes of Nucleophosmin (NPM1) Mutations Are Associated with Clinical Outcome in AML—A COG and SWOG Intergroup Collaboration. Blood 2016, 128, 285. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512.e9. [Google Scholar] [CrossRef] [Green Version]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.G.; Linhares, B.M.; Chen, D.; Jih, G.; Perkey, E.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [Google Scholar] [CrossRef] [Green Version]
- Kühn, M.W.M.; Song, E.; Feng, Z.; Sinha, A.; Chen, C.W.; Deshpande, A.J.; Cusan, M.; Farnoud, N.; Mupo, A.; Grove, C.; et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016, 6, 1166–1181. [Google Scholar] [CrossRef] [Green Version]
- Uckelmann, H.J.; Kim, S.M.; Wong, E.M.; Hatton, C.; Giovinazzo, H.; Gadrey, J.Y.; Krivtsov, A.V.; Rücker, F.G.; Döhner, K.; McGeehan, G.M.; et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 2020, 367, 586–590. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequence |
|---|---|
| NPM1_mutA_F | 5′ATCTCTGTCTGGCAGTGGAGGAAGTCTCTTTAAGAAAATAGTT 3′ |
| NPM1_mutA_R | 5′ACTGCCAGACAGAGATCTTGAATAGCCTCTTGGTCAGT 3′ |
| NPM1_mutB_F | 5′ATCTCTGCATGGCAGTGGAGGAAGTCTCTTTAAGAAAATAGTT 3′ |
| NPM1_mutB_R | 5′ACTGCCATGCAGAGATCTTGAATAGCCTCTTGGTCAGT 3′ |
| NPM1_mutD_F | 5′ATCTCTGCCTGGCAGTGGAGGAAGTCTCTTTAAGAAAATAGTT 3′ |
| NPM1_mutD_R | 5′ACTGCCAGGCAGAGATCTTGAATAGCCTCTTGGTCAGT 3′ |
| NPM1_mut5_F | 5′TGGCAGAGAATGGAGGAAGTCTCTTTAAGAAAATAGTTTAA 3′ |
| NPM1_mut5_R | 5′CTCCATTCTCTGCCAGAGATCTTGAATAGCCTCTTG 3′ |
| NPM1_mut7_F | 5′CAGTGCTTTTCAAAAGTCTCTTTAAGAAAATAGTTTAAACAA 3′ |
| NPM1_mut7_R | 5′ACTTTTGAAAAGCACTGCCAGAGATCTTGAATAGCCT 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tregnago, C.; Benetton, M.; Padrin, D.; Polato, K.; Borella, G.; Da Ros, A.; Marchetti, A.; Porcù, E.; Del Bufalo, F.; Mecucci, C.; et al. NPM1 Mutational Status Underlines Different Biological Features in Pediatric AML. Cancers 2021, 13, 3457. https://doi.org/10.3390/cancers13143457
Tregnago C, Benetton M, Padrin D, Polato K, Borella G, Da Ros A, Marchetti A, Porcù E, Del Bufalo F, Mecucci C, et al. NPM1 Mutational Status Underlines Different Biological Features in Pediatric AML. Cancers. 2021; 13(14):3457. https://doi.org/10.3390/cancers13143457
Chicago/Turabian StyleTregnago, Claudia, Maddalena Benetton, Davide Padrin, Katia Polato, Giulia Borella, Ambra Da Ros, Anna Marchetti, Elena Porcù, Francesca Del Bufalo, Cristina Mecucci, and et al. 2021. "NPM1 Mutational Status Underlines Different Biological Features in Pediatric AML" Cancers 13, no. 14: 3457. https://doi.org/10.3390/cancers13143457
APA StyleTregnago, C., Benetton, M., Padrin, D., Polato, K., Borella, G., Da Ros, A., Marchetti, A., Porcù, E., Del Bufalo, F., Mecucci, C., Locatelli, F., & Pigazzi, M. (2021). NPM1 Mutational Status Underlines Different Biological Features in Pediatric AML. Cancers, 13(14), 3457. https://doi.org/10.3390/cancers13143457