
Establishment of a Novel In Vitro Model of Endometriosis with Oncogenic KRAS and PIK3CA Mutations for Understanding the Underlying Biology and Molecular Pathogenesis

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Development of Immortalized Epithelial Cell Lines from Ovarian Endometriotic Tissue
2.2. Western Blot Analysis of pan-Cytokeratin, pan-AKT, pan-MAPK, p-MAPK, and p-AKT Expression in Immortalized Cells
2.3. Short Tandem Repeat (STR) Analysis
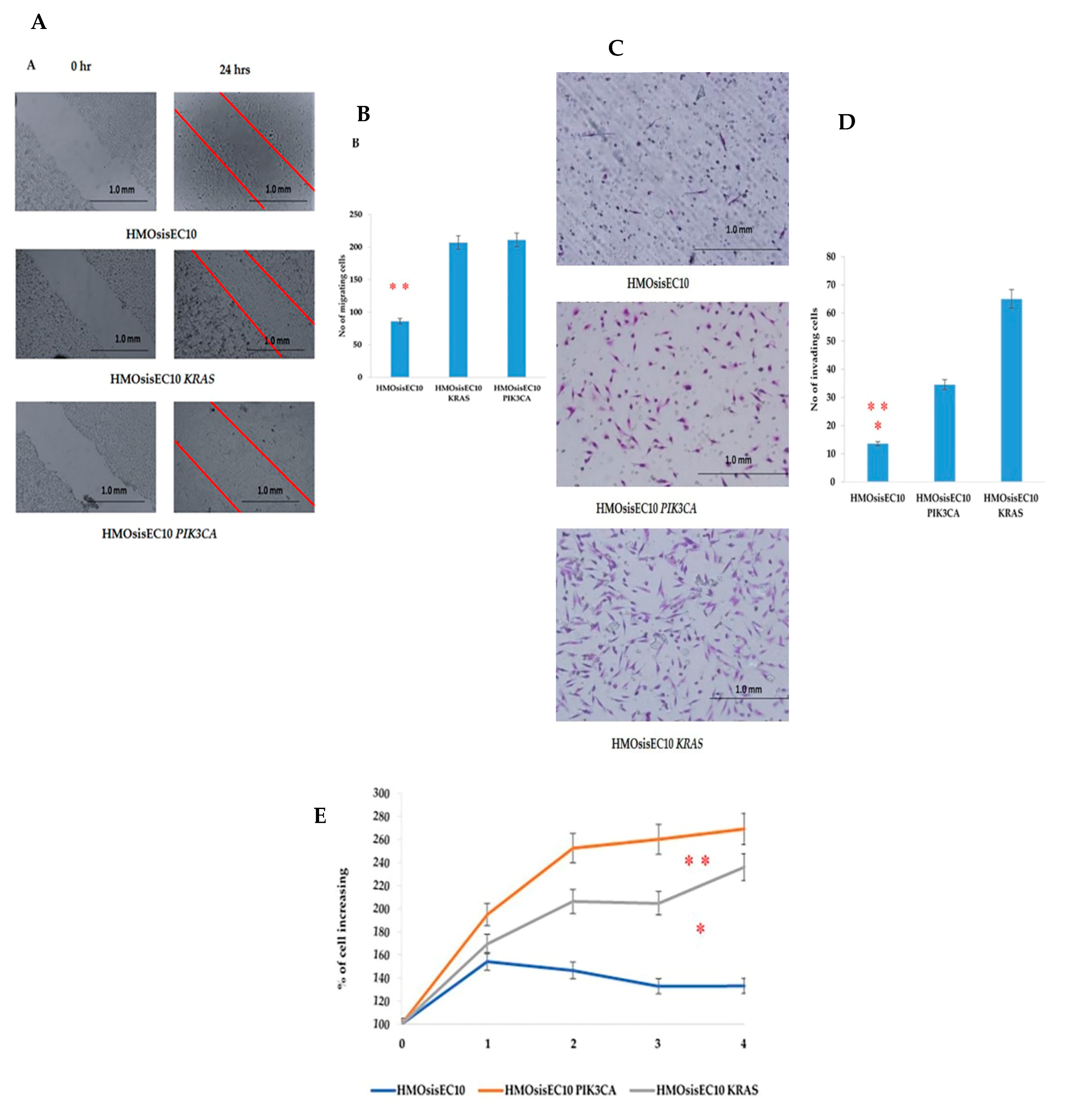
2.4. Migration, Invasion, Proliferation, and Anchorage-Independent Assays of Mutant Cells
2.5. Constitutive Effect of Tumorigenicity in HMOsisEC10 KRAS and HMOsisEC10 PIK3CA Mutant Cells
2.6. Lack of Other Genetic Mutations in Immortalized Epithelial Cells
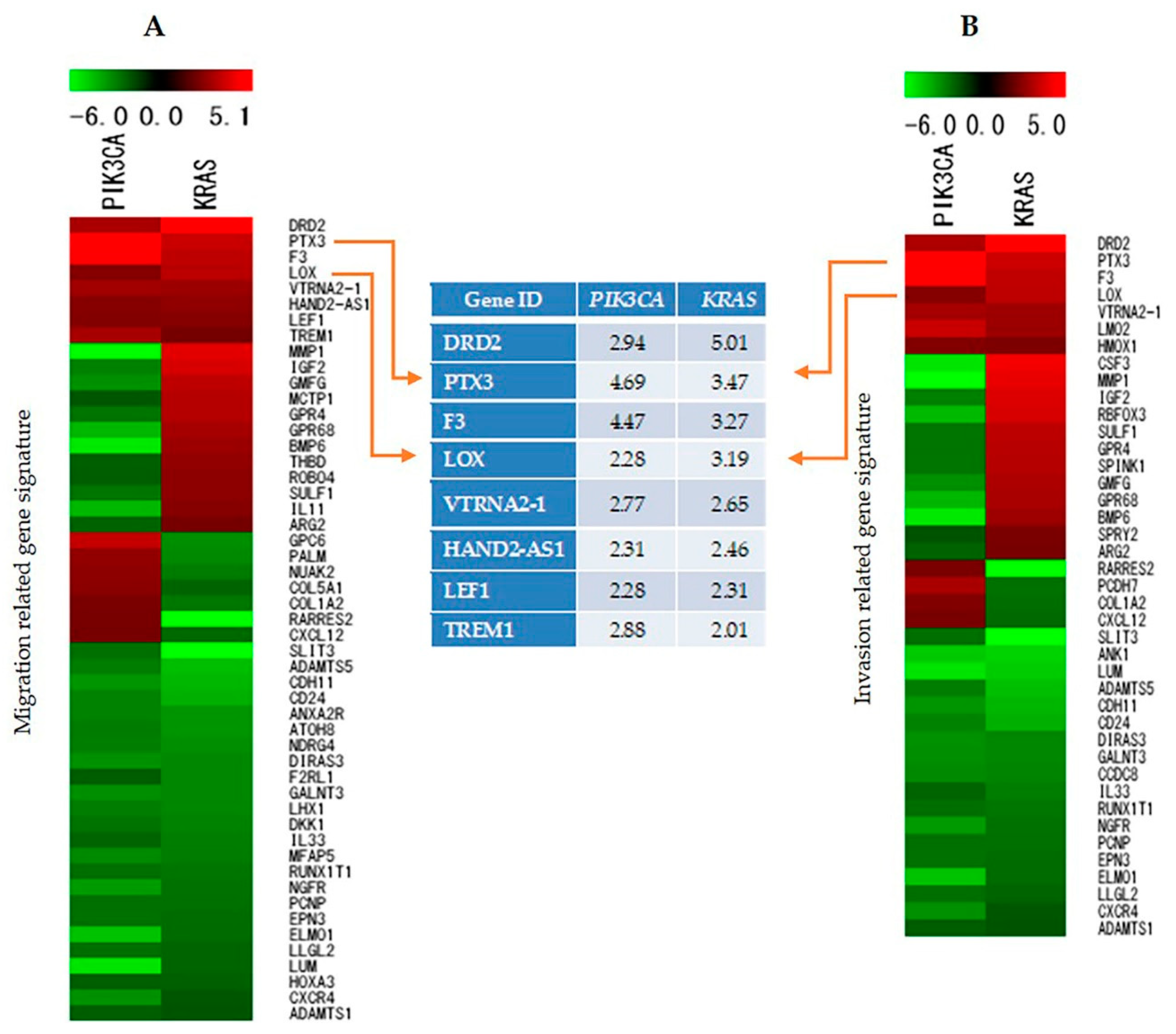
2.7. Identification and Analysis of Genes Involved in Cell Migration, Invasion, and Proliferation
2.8. Validation of Microarray Data by Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
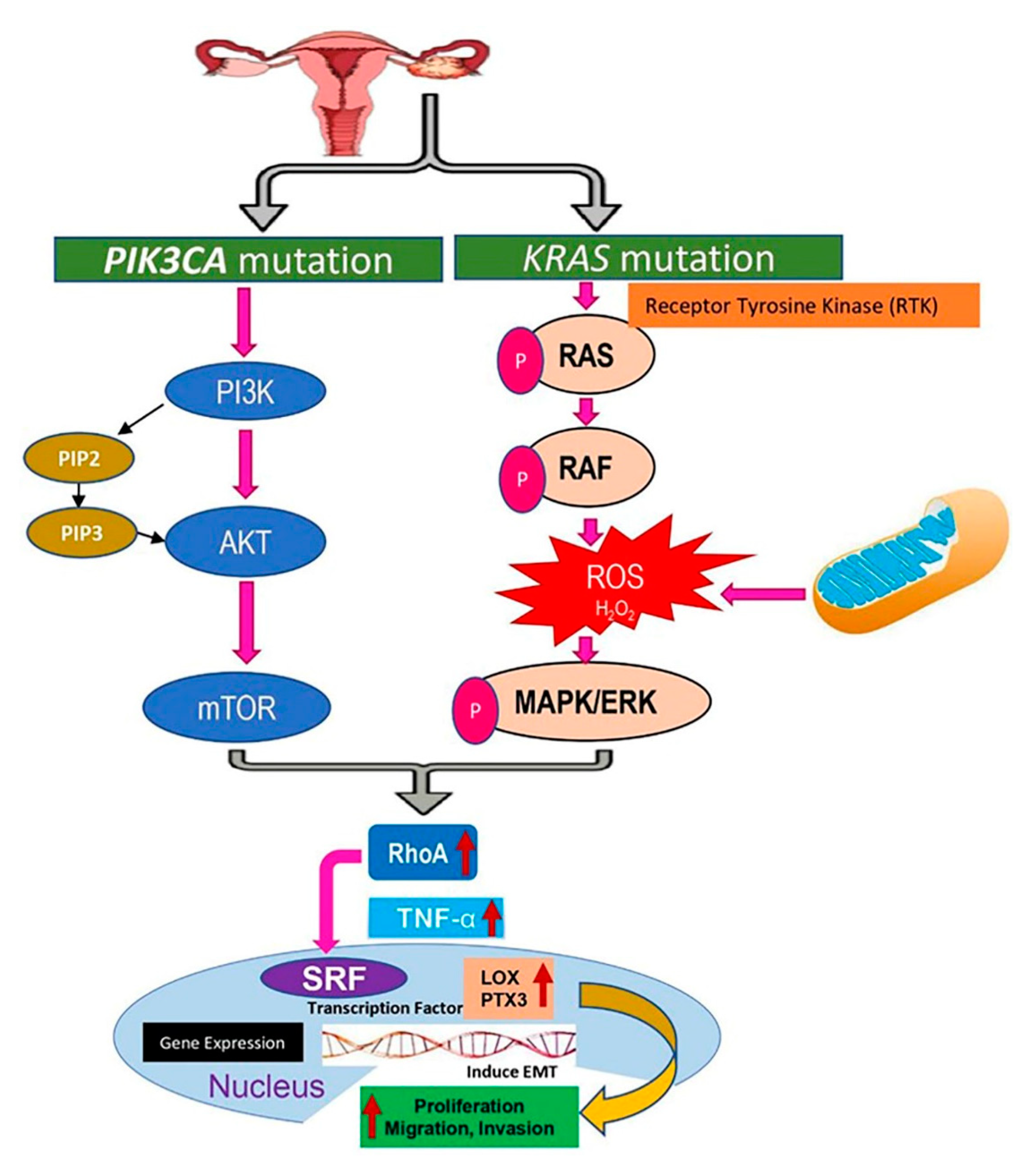
2.9. Signaling Pathways Targeting KRAS and PIK3CA Mutations in Endometriosis Due to the Upregulation of Lysyl Oxidase (LOX) and Pentraxin3 (PTX3)
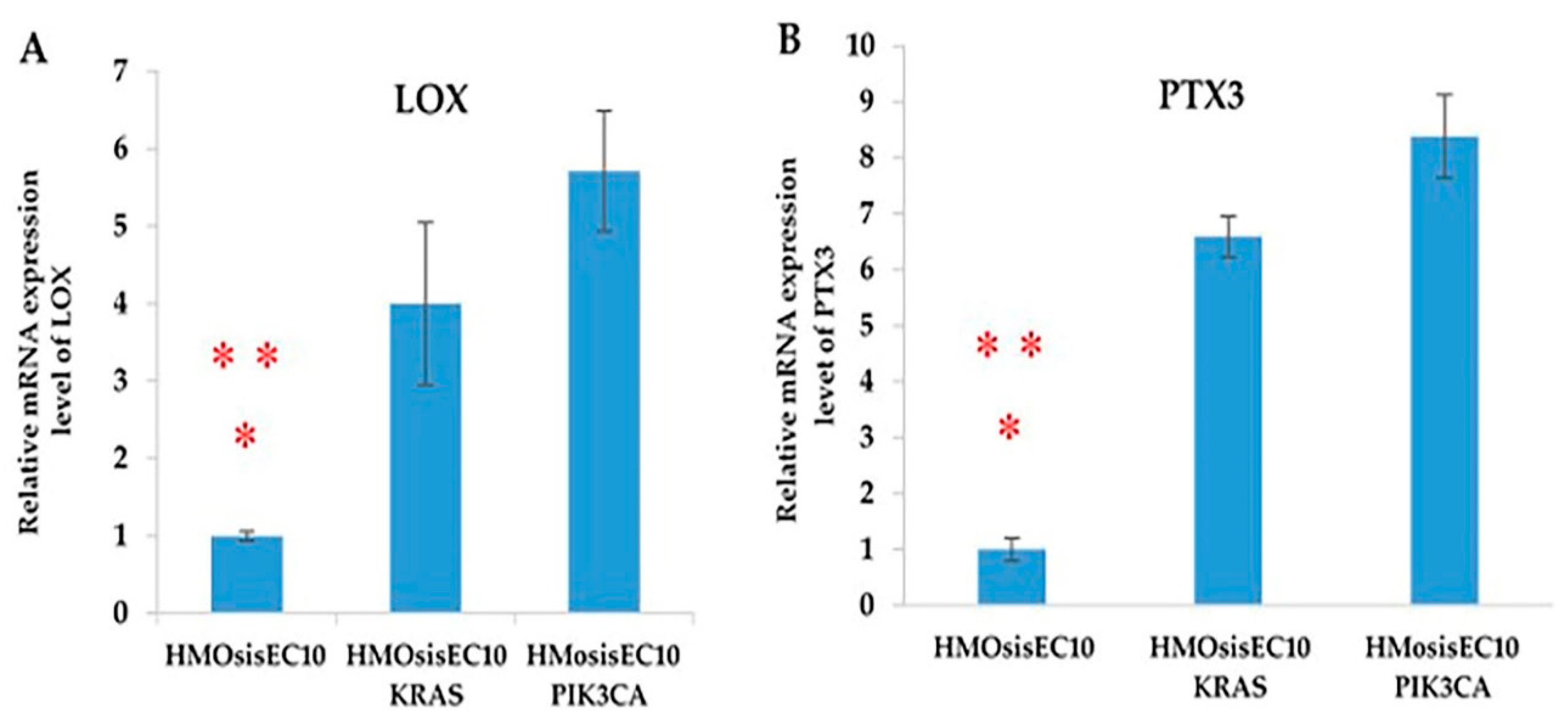
2.10. LOX and PTX3 mRNA Expression Levels Are Upregulated in Immortalized Endometriotic Mutant Epithelial Cells
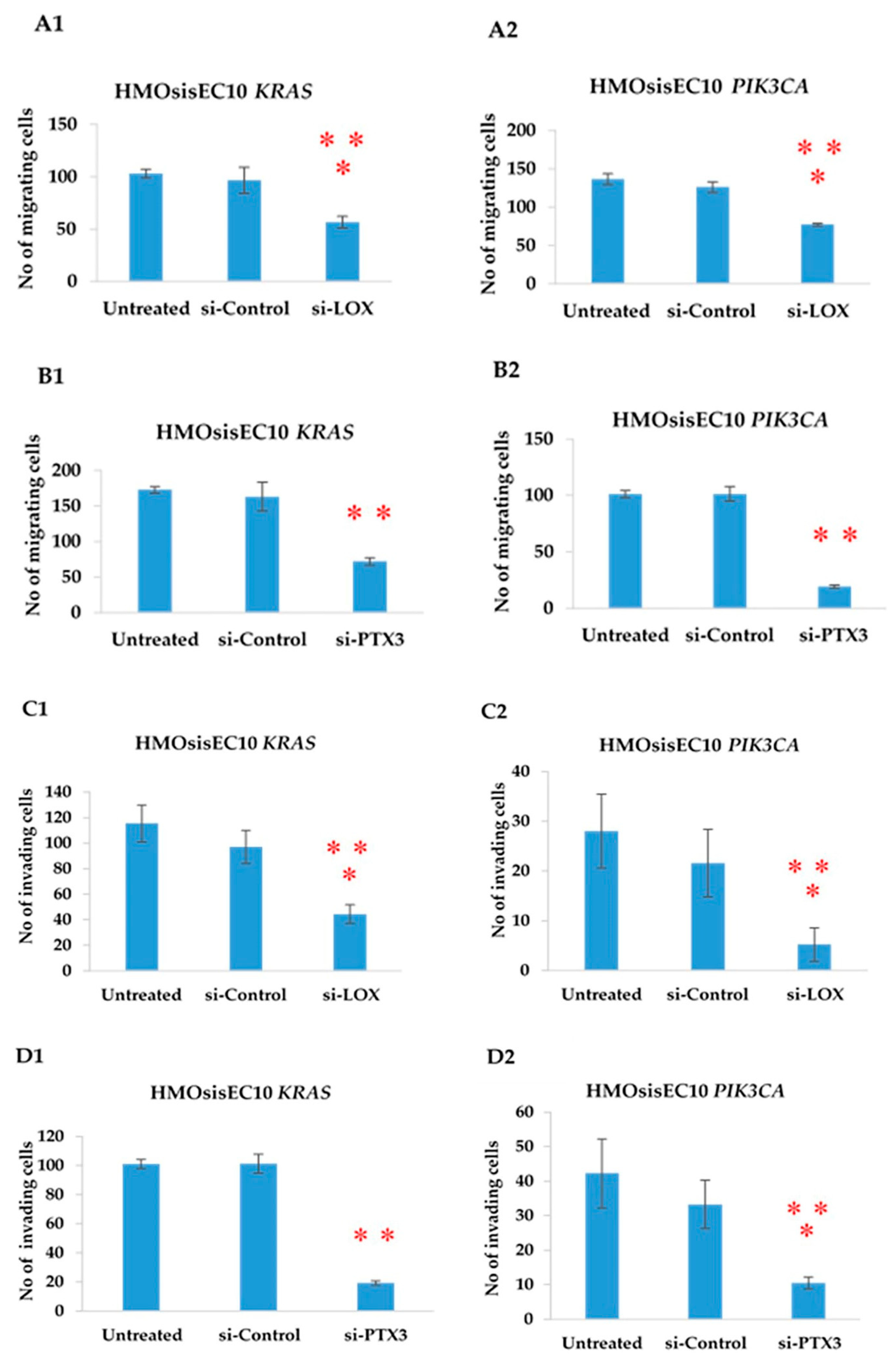
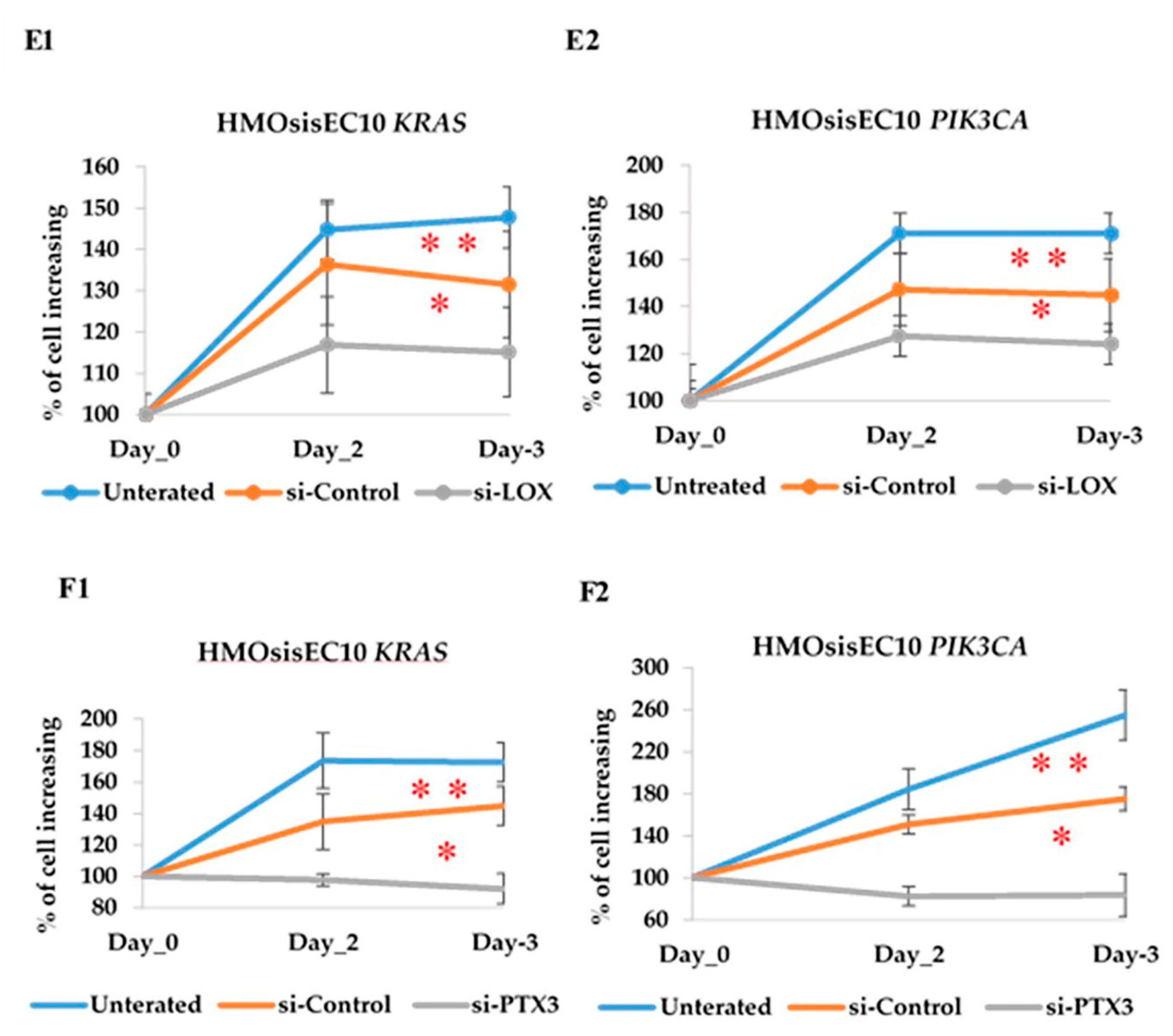
2.11. Constitutive Knockdown of LOX and PTX3 Leads to Decreased Migration, Invasion, and Proliferation in KRAS and PIK3CA Mutant Cell Lines
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Isolation and Purification of the Tissue Samples
4.3. Vector Construction and Cell Transfection
4.4. Cell Lines and Cell Culture
4.5. Western Blot Analysis
4.6. Immunocytochemistry
4.7. Short Tandem Repeat (STR) Analysis
4.8. Simulated Wound Healing Assay to Assess Cell Motility
4.9. Matrigel Invasion Assay
4.10. Cell Proliferation Assay
4.11. Anchorage Independent Assay
4.12. Nude Mice Xenograft
4.13. Whole-Exome Sequencing
4.14. Microarray Analysis
4.15. siRNA Transfection
4.16. Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| KRAS: | Kristen Rat Sarcoma |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| LOX | lysyl oxidase |
| PTX3 | Pentraxin3 |
| RT-PCR | Reverse Transcriptase Polymerase Chain Reaction |
| RhoA | Ras homolog family member A, |
| TNF-α | Tumor Necrotic Factor- α |
| SRF | Serum Response Factor |
| hTERT | humans Telomerase Reverse Transcriptase |
| CDK4 | Cyclin Dependent Kinase 4 |
| STR | Short Tandem Repeat |
| siRNA | small interfering RNA |
References
- Olive, D.L.; Pritts, E.A. Treatment of endometriosis. N. Engl. J. Med. 2001, 345, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Papadopoulos, N.; Ayhan, A.; Nazeran, T.M.; Noë, M.; Horlings, H.M.; Lum, A.; Jones, S.; Senz, J.; Seckin, T.; et al. Cancer associated mutation in Endometriosis without cancer. N. Engl. J. Med. 2017, 376, 1835–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoens, S.; Dunselman, G.; Dirksen, C.; Hummelshoj, L.; Bokor, A.; Brandes, I.; Brodszky, V.; Canis, M.; Colombo, G.L.; DeLeire, T.; et al. The burden of endometriosis: Costs and quality of life of women with endometriosis and treated in referral centers. Hum. Reprod. 2012, 27, 1292–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nnoaham, K.E.; Hummelshoj, L.; Webster, P.; d’Hooghe, T.; Nardone, F.C.; Jenkinson, C.; Phil, D.; Kennedy, S.H.; Zondervan, K.T. Impact of endometriosis on quality of life and work productivity: A multicenter study across ten countries. Fertil Steril. 2011, 96, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.J.; Lee, S.H.; Park, J.W.; Han, M.; Park, M.J.; Han, S.J. Dysfunctional signaling underlying endometriosis: Current state of knowledge. J. Mol. Endocrinol. 2018, 60, 97–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110. [Google Scholar]
- Vercellini, P.; Viganò, P.; Somigliana, E.; Fedele, L. Endometriosis: Pathogenesis and treatment. Nat. Rev. Endocrinol. 2014, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.P.; Hummelshoj, L.; Adamson, G.D.; Keckstein, J.; Taylor, H.S.; Abrao, M.S.; Bush, D.; Kiesel, L.; Tamimi, R.; Sharpe-Timms, K.L.; et al. World Endometriosis Society consensus on the classification of endometriosis. Hum. Reprod. 2017, 32, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Tsunoda, H.; Nishida, M.; Morishita, Y.; Takimoto, Y.; Kubo, T.; Noguchi, M. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: Possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000, 60, 7052–7056. [Google Scholar] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutation in endometriosis- Associated Ovarian Carcinoma. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, A.L.; Thorup, K.; Knudsen, U.B.; Munk, T.; Rosbach, H.; Poulsen, J.B.; Guldberg, P.; Martensen, P.M. Oncogenic events associated with endometrial and ovarian cancer are rare endometriosis. Mol. Hum. Reprod. 2011, 17, 758–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Kotani, Y.; Nakai, H.; Matsumura, N. Endometriosis-Associated Ovarian Cancer: The Origin and Targeted Therapy. Cancers 2020, 12, 1676. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, H.; Yoshikawa, H.; Onda, T.; Yasugi, T.; Sakamoto, A.; Taketani, Y. Prevalence of ovarian endometriosis in epithelial ovarian cancer. Int. J. Gynaecol. Obstet. 1997, 59, 245–250. [Google Scholar] [CrossRef]
- Wei, J.J.; William, J.; Bulun, S. Endometriosis and ovarian cancer: A review of clinical, pathologic, and molecular aspects. Int. J. Gynecol. Pathol. 2011, 30, 553–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal expansion and diversification of cancer associated mutation in endometriosis and normal endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Adachi, S.; Kase, H.; Motoyama, T.; Inoue, I.; Enomoto, T. Different mutation profiles between epithelium and stroma in endometriosis and normal endometrium. Hum. Reprod. 2019, 34, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Cruz Diaz, L.A.; Yoshihara, K.; Nakaoka, H.; Yachida, N.; Motoyama, T.; Inoue, I.; Enomoto, T. Clonal lineage from normal endometrium to ovarian clear cell carcinoma through ovarian endometriosis. Cancer Sci. 2020, 111, 3000–3009. [Google Scholar] [CrossRef] [PubMed]
- Lac, V.; Verhoef, L.; Aguirre-Hernandez, R.; Nazeran, T.M.; Tessier-Cloutier, B.; Praetorius, T.; Orr, N.L.; Noga, H.; Lum, A.; Khattra, J.; et al. Iatrogenic endometriosis harbors somatic cancer-driver mutations. Hum. Reprod. 2019, 34, 69–78. [Google Scholar] [CrossRef]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Hirota, Y.; Ueno, T.; Fukui, Y.; Yoshida, E.; Hayashi, T.; Kojima, S.; Takeyama, R.; Hashimoto, T.; Kiyono, T.; et al. Uterine adenomyosis is an oligoclonal disorder associated with KRAS mutations. Nat. Commun. 2019, 10, 5785. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H. Somatic driver mutations in endometriosis as possible regulators of fibrogenesis (Review). World Acad. Sci. J. 2019, 12, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, K.; Kiyono, T.; Okamura, K.; Uezumi, M.; Goto, Y.; Yasumoto, S.; Shimizu, S.; Hashimoto, N. CDK4 and Cyclin D1 Allow Human Myogenic Cells to Recapture Growth Property Without Compromising Differentiation Potential. Gene Ther. 2011, 18, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Benvenuti, S.; Frattini, M.; Arena, S.; Zanon, C.; Cappelletti, V.; Coradini, D.; Daidone, M.G.; Pilotti, S.; Pierotti, M.A.; Bardelli, A. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat. 2008, 29, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol. Onchol. 2013, 7, 165–177. [Google Scholar] [CrossRef]
- Gutierrez, E.; Cahatol, I.; Bailey, C.A.R.; Lafargue, A.; Zhang, N.; Song, Y.; Tian, H.; Zhang, Y.; Chan, R.; Gu, K.; et al. Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes. Cancers 2019, 11, 818. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. RhoA, RhoB and RhoC have different roles in cancer cell migration. J. Microsc. 2013, 251, 242–249. [Google Scholar] [CrossRef]
- Mong, P.Y.; Petrulio, C.; Kaufman, H.L.; Wang, Q. Activation of Rho kinase by TNF-alpha is required for JNK activation in human pulmonary microvascular endothelial cells. J. Immunol. 2008, 180, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Berenjeno, I.M.; Núñez, F.; Bustelo, X.R. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene 2007, 26, 4295–4305. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.W. Cancer driver mutations in endometriosis: Variations on the major theme of fibrogenesis. Reprod. Med. Biol. 2018, 16, 369–397. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, J.; Okuda, T.; Sekizawa, A.; Amemiya, S.; Saito, H.; Okai, T.; Kushima, M.; Tachikawa, T. K-ras mutation may promote carcinogenesis of endometriosis leading to ovarian clear cell carcinoma. Med. Electron. Microsc. 2004, 37, 188–192. [Google Scholar] [CrossRef]
- Rachagani, S.; Senapati, S.; Chakraborty, S.; Ponnusamy, M.P.; Kumar, S.; Smith, L.M.; Jain, M.; Batra, S.K. Activated KrasG¹²D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br. J. Cancer 2011, 104, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, Y.; Diaz, L.A., Jr.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Lian, S.T.; Chen, Y.F.; Yang, Y.C.; Chen, L.T.; Lee, K.T.; Huang, T.J.; Lin, S.R. Unique K-ras Mutational Pattern in Pancreatic Adenocarcinoma from Taiwanese Patients. Cancer Lett. 2002, 180, 153–158. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous Oncogenic K-ras(G12D) Stimulates Proliferation and Widespread Neoplastic and Developmental Defects. Cancer Cell. 2004, 5, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Dawson, A.; Fernandez, M.L.; Anglesio, M.; Yong, P.J.; Carey, M.S. Endometriosis and endometriosis-associated cancers: New insights into the molecular mechanisms of ovarian cancer development. Ecancermedicalscience 2018, 12, 803. [Google Scholar] [CrossRef] [Green Version]
- Heaps, J.M.; Nieberg, R.K.; Berek, J.S. Malignant neoplasms arising in endometriosis. Obstet Gynecol. 1990, 75, 1023–1028. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer --Three Strikes and You’re Out. N. Engl. J. Med. 2015, 373, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, G.; Liang, Z.; Mei, Z.; Wu, T.; Cui, A.; Liu, C.; Cui, L. Lysyl oxidase: A colorectal cancer biomarker of lung and hepatic metastasis. Thorac. Cancer 2018, 9, 785–793. [Google Scholar] [CrossRef]
- Ruiz, L.A.; Báez-Vega, P.M.; Ruiz, A.; Peterse, D.P.; Monteiro, J.B.; Bracero, N.; Beauchamp, P.; Fazleabas, A.T.; Flores, I. Dysregulation of Lysyl Oxidase Expression in Lesions and Endometrium of Women with Endometriosis. Reprod. Sci. 2015, 22, 1496–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalikawe, R.; Baba, Y.; Nomoto, D.; Okadome, K.; Miyake, K.; Eto, K.; Hiyoshi, Y.; Nagai, Y.; Iwatsuki, M.; Ishimoto, T.; et al. Lysyl oxidase impacts disease outcomes and correlates with global DNA hypomethylation in esophageal cancer. Cancer Sci. 2019, 110, 3727–3737. [Google Scholar] [CrossRef]
- Song, T.; Wang, C.; Guo, C.; Liu, Q.; Zheng, X. Pentraxin 3 overexpression accelerated tumor metastasis and indicated poor prognosis in hepatocellular carcinoma via driving epithelial-mesenchymal transition. J. Cancer 2018, 9, 2650–2658. [Google Scholar] [CrossRef]
- Moon, H.J.; Finney, J.; Xu, L.; Moore, D.; Welch, D.R.; Mure, M. MCF-7 cells expressing nuclear associated lysyl oxidase-like 2 (LOXL2) exhibit an epithelial-to-mesenchymal transition (EMT) phenotype and are highly invasive in vitro. J. Biol. Chem. 2013, 288, 30000–30008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.H.; Zhuang, M.K.; Xie, W.H.; Du, F.; Huang, Y.H.; Chen, Z.X.; Chen, F.L.; Wang, X.Z. 12-Lipoxygenase promotes epithelial–mesenchymal transition via the Wnt/β-catenin signaling pathway in gastric cancer cells. Oncol. Targets Ther. 2019, 12, 5551–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Donato, M.; Petrillo, M.; Martinelli, E.; Filippetti, F.; Zannoni, G.F.; Scambia, G.; Gallo, D. Uncovering the role of nuclear Lysyl oxidase (LOX) in advanced high grade serous ovarian cancer. Gynecol. Oncol. 2017, 146, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Rathore, M.; Girard, C.; Ohanna, M.; Tichet, M.; Ben Jouira, R.; Garcia, E.; Larbret, F.; Gesson, M.; Audebert, S.; Lacour, J.P.; et al. Cancer Cell-Derived Long Pentraxin 3 (PTX3) Promotes Melanoma Migration through a Toll-Like Receptor 4 (TLR4)/NF-κB Signaling Pathway. Oncogene 2019, 38, 5873–5889. [Google Scholar] [CrossRef]
- Ying, T.H.; Lee, C.H.; Chiou, H.L.; Yang, S.F.; Lin, C.L.; Hung, C.H.; Tsai, J.P.; Hsieh, Y.H. Knockdown of Pentraxin 3 suppresses tumorigenicity and metastasis of human cervical cancer cells. Sci. Rep. 2016, 6, 29385. [Google Scholar] [CrossRef] [PubMed]
- Vercellini, P.; Somigliana, E.; Viganò, P.; De Matteis, S.; Barbara, G.; Fedele, L. The effect of second-line surgery on reproductive performance of women with recurrent endometriosis: A systematic review. Acta Obstet. Gynecol. Scand. 2009, 88, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Elnashar, A. Emerging treatment of endometriosis. Middle East Fertil. Soc. J. 2015, 20, 61–69. [Google Scholar] [CrossRef]
- Lee, I.I.; Kim, J.J. Influence of AKT on progesterone action in endometrial diseases. Biol. Reprod. 2014, 91, 63. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.M.; Coutinho, L.M.; Vannuccini, S.; Batteux, F.; Chapron, C.; Petraglia, F. Progesterone receptor ligands for the treatment of endometriosis: The mechanisms behind therapeutic success and failure. Hum. Reprod. Update 2020, 26, 565–585. [Google Scholar] [CrossRef] [PubMed]
- Bono, Y.; Kyo, S.; Takakura, M.; Maida, Y.; Mizumoto, Y.; Nakamura, M.; Nomura, K.; Kiyono, T.; Inoue, M. Creation of immortalized epithelial cells from ovarian endometrioma. Br. J. Cancer 2012, 106, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Miyoshi, H.; Blömer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a Self-Inactivating Lentivirus Vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Miyazaki, K.; Kanzaki, A.; Fukumoto, M.; Takebayashi, Y. Expression and Cisplatin Sensitivity of Copper-Transporting P-type Adenosine Triphosphates (ATP7B) in Human Solid Carcinoma Cell Lines. Oncol. Rep. 2001, 8, 1285–1287. [Google Scholar] [PubMed]
- Nakamura, K.; Aimono, E.; Tanishima, S.; Imai, M.; Nagatsuma, A.K.; Hayashi, H.; Yoshimura, Y.; Nakayama, K.; Kyo, S.; Nishihara, H. Intratumoral Genomic Heterogeneity May Hinder Precision Medicine Strategies in Patients with Serous Ovarian Carcinoma. Diagnostics 2020, 10, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Primer | Type | Sequence (5′-3′) |
|---|---|---|
| LOX | Forward | TGCCAGTGGATTGATATTAC |
| Reverse | TACGGTGAAATTGTGCAGCC | |
| PTX3 | Forward | GCATCTCCTTGCGATTCTGTT |
| Reverse | CATTCCGAGTGCTCCTGACC | |
| GAPDH | Forward | ACGGGAAGCTTGTCATCAAT |
| Reverse | TGGACTCCACGACGTACTCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hossain, M.M.; Nakayama, K.; Shanta, K.; Razia, S.; Ishikawa, M.; Ishibashi, T.; Yamashita, H.; Sato, S.; Iida, K.; Kanno, K.; et al. Establishment of a Novel In Vitro Model of Endometriosis with Oncogenic KRAS and PIK3CA Mutations for Understanding the Underlying Biology and Molecular Pathogenesis. Cancers 2021, 13, 3174. https://doi.org/10.3390/cancers13133174
Hossain MM, Nakayama K, Shanta K, Razia S, Ishikawa M, Ishibashi T, Yamashita H, Sato S, Iida K, Kanno K, et al. Establishment of a Novel In Vitro Model of Endometriosis with Oncogenic KRAS and PIK3CA Mutations for Understanding the Underlying Biology and Molecular Pathogenesis. Cancers. 2021; 13(13):3174. https://doi.org/10.3390/cancers13133174
Chicago/Turabian StyleHossain, Mohammad Mahmud, Kentaro Nakayama, Kamrunnahar Shanta, Sultana Razia, Masako Ishikawa, Tomoka Ishibashi, Hitomi Yamashita, Seiya Sato, Kouji Iida, Kosuke Kanno, and et al. 2021. "Establishment of a Novel In Vitro Model of Endometriosis with Oncogenic KRAS and PIK3CA Mutations for Understanding the Underlying Biology and Molecular Pathogenesis" Cancers 13, no. 13: 3174. https://doi.org/10.3390/cancers13133174