
Aspirin-Triggered Resolvin D1 Reduces Proliferation and the Neutrophil to Lymphocyte Ratio in a Mutant KRAS-Driven Lung Adenocarcinoma Model

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Immunohistochemical and Immunofluorescence Staining for Neutrophils, Macrophages and Lymphocytes
2.3. TCGA and Oncolnc Database Analysis
2.4. Gene Expression Analysis by RTqPCR and Kras Genotyping
2.5. Quantification of SAA in Serum
2.6. Kras Mouse Model
2.7. Statistical Analysis
3. Results
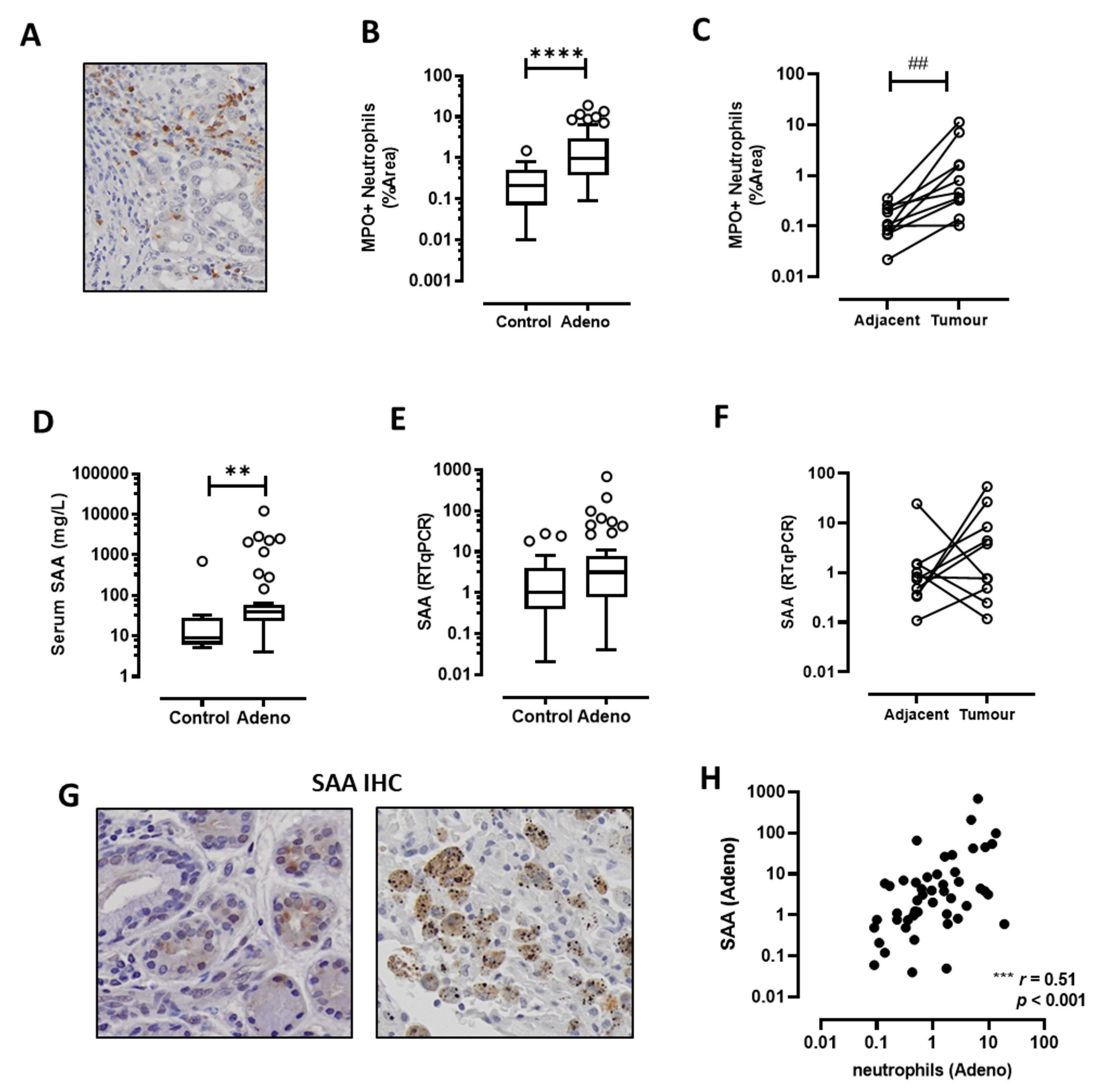
3.1. TANs Accumulate in Adenocarcinoma Biopsies and Correlate with SAA Expression
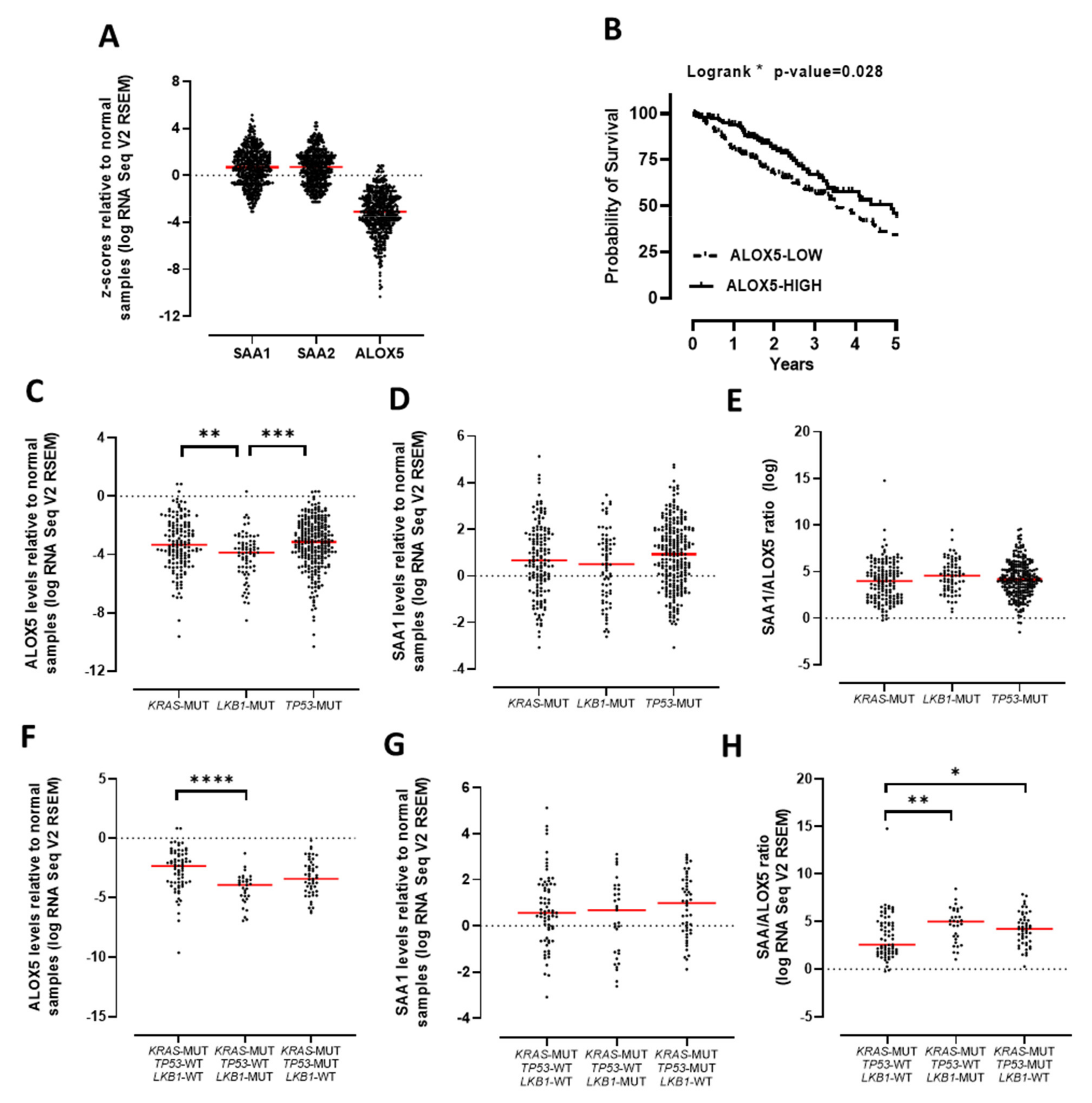
3.2. The SAA/ALOX5 Ratio Is Increased in KRAS Mutated Lung Adenocarcinoma
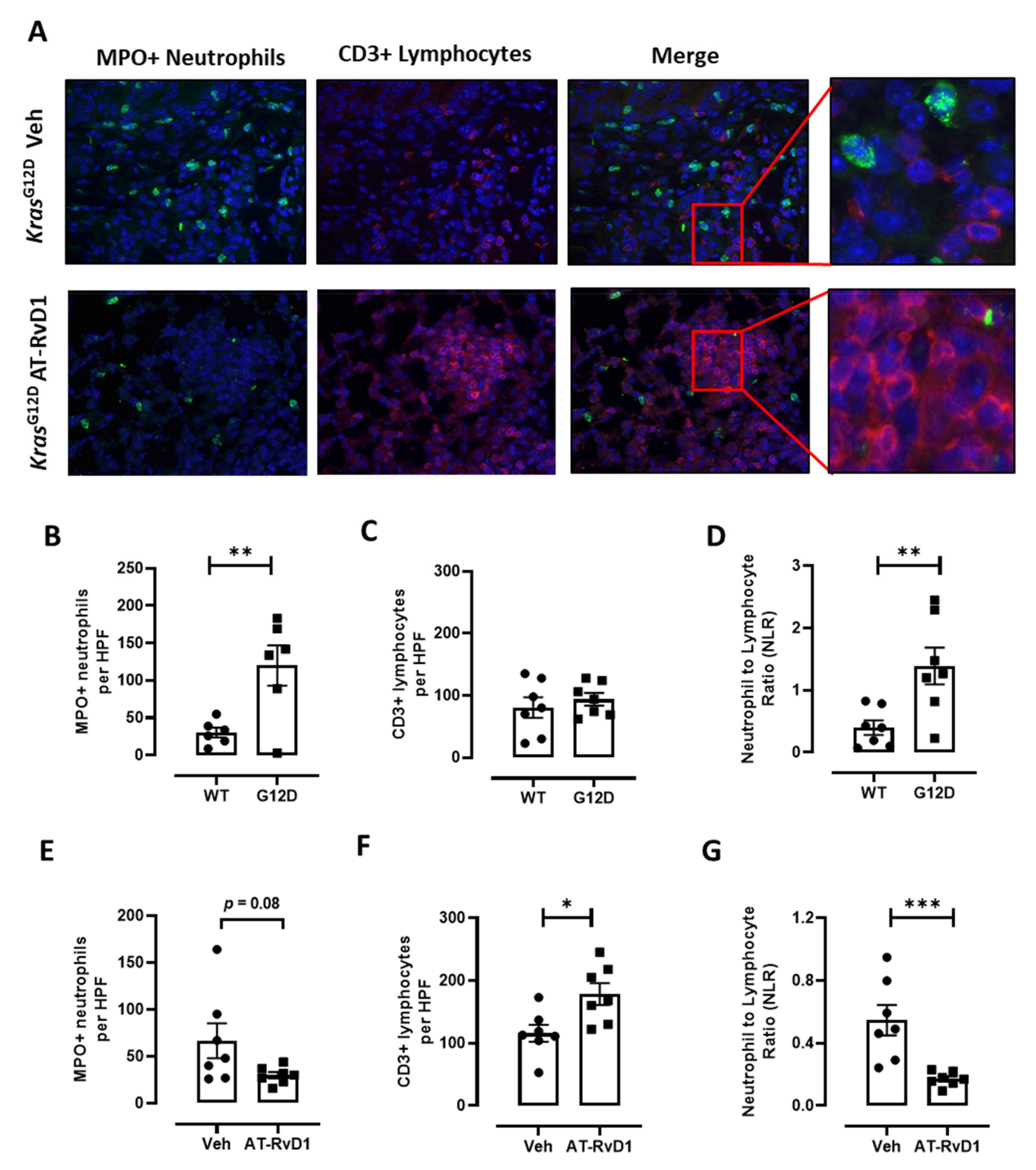
3.3. AT-RvD1 Reduces Tumour Growth and the NLR in KrasG12D-Induced Lung Adenocarcinoma
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Cha, Y.I.; Dubois, R.N. NSAIDs and Cancer Prevention: Targets Downstream of COX-2. Annu. Rev. Med. 2007, 58, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Edelman, M.J.; Wang, X.; Hodgson, L.; Cheney, R.T.; Baggstrom, M.Q.; Thomas, S.P.; Gajra, A.; Bertino, E.; Reckamp, K.L.; Molina, J.; et al. Phase III Randomized, Placebo-Controlled, Double-Blind Trial of Celecoxib in Addition to Standard Chemotherapy for Advanced Non–Small-Cell Lung Cancer with Cyclooxygenase-2 Overexpression: CALGB 30801 (Alliance). J. Clin. Oncol. 2017, 35, 2184–2192. [Google Scholar] [CrossRef]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.-L. Resolvins. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Bozinovski, S.; Vlahos, R.; Anthony, D.; McQualter, J.; Anderson, G.; Irving, L.; Steinfort, D. COPD and squamous cell lung cancer: Aberrant inflammation and immunity is the common link. Br. J. Pharmacol. 2015, 173, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Bozinovski, S.; Anthony, D.; Anderson, G.P.; Irving, L.B.; Levy, B.D.; Vlahos, R. Treating neutrophilic inflammation in COPD by targeting ALX/FPR2 resolution pathways. Pharmacol. Ther. 2013, 140, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2017, 215, 115–140. [Google Scholar] [CrossRef]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Anthony, D.; Seow, H.J.; Uddin, M.; Thompson, M.; Dousha, L.; Vlahos, R.; Irving, L.B.; Levy, B.D.; Anderson, G.P.; Bozinovski, S. Serum Amyloid A Promotes Lung Neutrophilia by Increasing IL-17A Levels in the Mucosa and γδ T Cells. Am. J. Respir. Crit. Care Med. 2013, 188, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Bozinovski, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.-S.; Wark, P.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum amyloid A opposes lipoxin A4 to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar] [CrossRef] [Green Version]
- De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-M.; Kim, E.-K.; Seo, H.; Jeon, I.; Chae, M.-J.; Park, Y.-J.; Song, B.; Kim, Y.-S.; Kim, Y.-J.; Ko, H.-J.; et al. Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid-derived suppressor cells via TLR2-dependent STAT3 activation. Eur. J. Immunol. 2014, 44, 1672–1684. [Google Scholar] [CrossRef]
- Guthrie, G.J.; Charles, K.A.; Roxburgh, C.S.; Horgan, P.G.; McMillan, D.; Clarke, S.J. The systemic inflammation-based neutrophil–lymphocyte ratio: Experience in patients with cancer. Crit. Rev. Oncol. 2013, 88, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Hofman, V.; Ortholan, C.; Bonnetaud, C.; Coëlle, C.; Mouroux, J.; Hofman, P. Predictive clinical outcome of the intratumoral CD66b-positive neutrophil-to-CD8-positive T-cell ratio in patients with resectable nonsmall cell lung cancer. Cancer 2011, 118, 1726–1737. [Google Scholar] [CrossRef]
- Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannitamby, A.; Seow, H.J.; Anderson, G.; Vlahos, R.; Thompson, M.; Steinfort, D.; Irving, L.B.; Bozinovski, S. Tumour-associated neutrophils and loss of epithelial PTEN can promote corticosteroid-insensitive MMP-9 expression in the chronically inflamed lung microenvironment. Thorax 2017, 72, 1140–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannitamby, A.; Hendry, S.; Makadia, T.; Danks, J.; Slavin, J.; Irving, L.; Steinfort, D.; Bozinovski, S. A Novel Approach to Detect Programed Death Ligand 1 (PD-L1) Status and Multiple Tumor Mutations Using a Single Non–Small-Cell Lung Cancer (NSCLC) Bronchoscopy Specimen. J. Mol. Diagn. 2019, 21, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janku, F.; Huang, H.J.; Fujii, T.; Shelton, D.N.; Madwani, K.; Fu, S.; Tsimberidou, A.M.; Piha-Paul, S.A.; Wheler, J.J.; Zinner, R.G.; et al. MultiplexKRASG12/G13 mutation testing of unamplified cell-free DNA from the plasma of patients with advanced cancers using droplet digital polymerase chain reaction. Ann. Oncol. 2017, 28, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM 17 selectively activates the IL -6 trans-signaling/ERK MAPK axis in KRAS -addicted lung cancer. EMBO Mol. Med. 2019, 11, e9976. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [Green Version]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [Green Version]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Esteinhilber, D.; Fischer, A.S.; Emetzner, J.; Steinbrink, S.D.; Eroos, J.; Eruthardt, M.; Maier, T.J. 5-Lipoxygenase: Underappreciated Role of a Pro-Inflammatory Enzyme in Tumorigenesis. Front. Pharmacol. 2010, 1, 143. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, X.; Song, G.; Luo, D. Prognostic significance of LKB1 promoter methylation in cutaneous malignant melanoma. Oncol. Lett. 2017, 14, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Rådmark, O.; Samuelsson, B. 5-Lipoxygenase: Mechanisms of regulation. J. Lipid Res. 2009, 50, S40–S45. [Google Scholar] [CrossRef] [Green Version]
- Uhl, J.; Klan, N.; Rose, M.; Entian, K.-D.; Werz, O.; Steinhilber, D. The 5-Lipoxygenase Promoter Is Regulated by DNA Methylation. J. Biol. Chem. 2002, 277, 4374–4379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieshober, L.; Graw, S.; Barnett, M.J.; Thornquist, M.D.; Goodman, G.E.; Chen, C.; Koestler, D.C.; Marsit, C.; Doherty, J.A. Methylation-derived Neutrophil-to-Lymphocyte Ratio and Lung Cancer Risk in Heavy Smokers. Cancer Prev. Res. 2018, 11, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poczobutt, J.M.; Nguyen, T.T.; Hanson, D.; Li, H.; Sippel, T.R.; Weiser-Evans, M.C.M.; Gijon, M.; Murphy, R.C.; Nemenoff, R.A. Deletion of 5-Lipoxygenase in the Tumor Microenvironment Promotes Lung Cancer Progression and Metastasis through Regulating T Cell Recruitment. J. Immunol. 2015, 196, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.L.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filep, J.G. Biasing the lipoxin A4/formyl peptide receptor 2 pushes inflammatory resolution. Proc. Natl. Acad. Sci. USA 2013, 110, 18033–18034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Blackall, M.; Sominsky, L.; Spencer, S.J.; Vlahos, R.; Churchill, M.; Bozinovski, S. Increased hypothalamic microglial activation after viral-induced pneumococcal lung infection is associated with excess serum amyloid A production. J. Neuroinflamm. 2018, 15, 200. [Google Scholar] [CrossRef]
- Yang, M.; Liu, F.; Higuchi, K.; Sawashita, J.; Fu, X.; Zhang, L.; Zhang, L.; Fu, L.; Tong, Z.; Higuchi, K. Serum amyloid A expression in the breast cancer tissue is associated with poor prognosis. Oncotarget 2016, 7, 35843–35852. [Google Scholar] [CrossRef] [Green Version]
- Hagihara, K.; Nishikawa, T.; Isobe, T.; Song, J.; Sugamata, Y.; Yoshizaki, K. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem. Biophys. Res. Commun. 2004, 314, 363–369. [Google Scholar] [CrossRef]
- Ancrile, B.; Lim, K.-H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cai, L.; Wang, H.; Wu, P.; Gu, W.; Chen, Y.; Hao, H.; Tang, K.; Yi, P.; Liu, M.; et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011, 30, 3887–3899. [Google Scholar] [CrossRef] [Green Version]
- Mattoscio, D.; Isopi, E.; Lamolinara, A.; Patruno, S.; Medda, A.; De Cecco, F.; Chiocca, S.; Iezzi, M.; Romano, M.; Recchiuti, A. Resolvin D1 reduces cancer growth stimulating a protective neutrophil-dependent recruitment of anti-tumor monocytes. J. Exp. Clin. Cancer Res. 2021, 40, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kramer, P.; Prichard, L.; Chacko, B.; Ravi, S.; Overton, E.T.; Heath, S.L.; Darley-Usmar, V. Inhibition of the lymphocyte metabolic switch by the oxidative burst of human neutrophils. Clin. Sci. 2015, 129, 489–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 32. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannitamby, A.; Saad, M.I.; Aloe, C.; Wang, H.; Kumar, B.; Vlahos, R.; Selemidis, S.; Irving, L.; Steinfort, D.; Jenkins, B.J.; et al. Aspirin-Triggered Resolvin D1 Reduces Proliferation and the Neutrophil to Lymphocyte Ratio in a Mutant KRAS-Driven Lung Adenocarcinoma Model. Cancers 2021, 13, 3224. https://doi.org/10.3390/cancers13133224
Vannitamby A, Saad MI, Aloe C, Wang H, Kumar B, Vlahos R, Selemidis S, Irving L, Steinfort D, Jenkins BJ, et al. Aspirin-Triggered Resolvin D1 Reduces Proliferation and the Neutrophil to Lymphocyte Ratio in a Mutant KRAS-Driven Lung Adenocarcinoma Model. Cancers. 2021; 13(13):3224. https://doi.org/10.3390/cancers13133224
Chicago/Turabian StyleVannitamby, Amanda, Mohamed I. Saad, Christian Aloe, Hao Wang, Beena Kumar, Ross Vlahos, Stavros Selemidis, Louis Irving, Daniel Steinfort, Brendan J. Jenkins, and et al. 2021. "Aspirin-Triggered Resolvin D1 Reduces Proliferation and the Neutrophil to Lymphocyte Ratio in a Mutant KRAS-Driven Lung Adenocarcinoma Model" Cancers 13, no. 13: 3224. https://doi.org/10.3390/cancers13133224