
Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma




Abstract
:Simple Summary
Abstract
1. Introduction
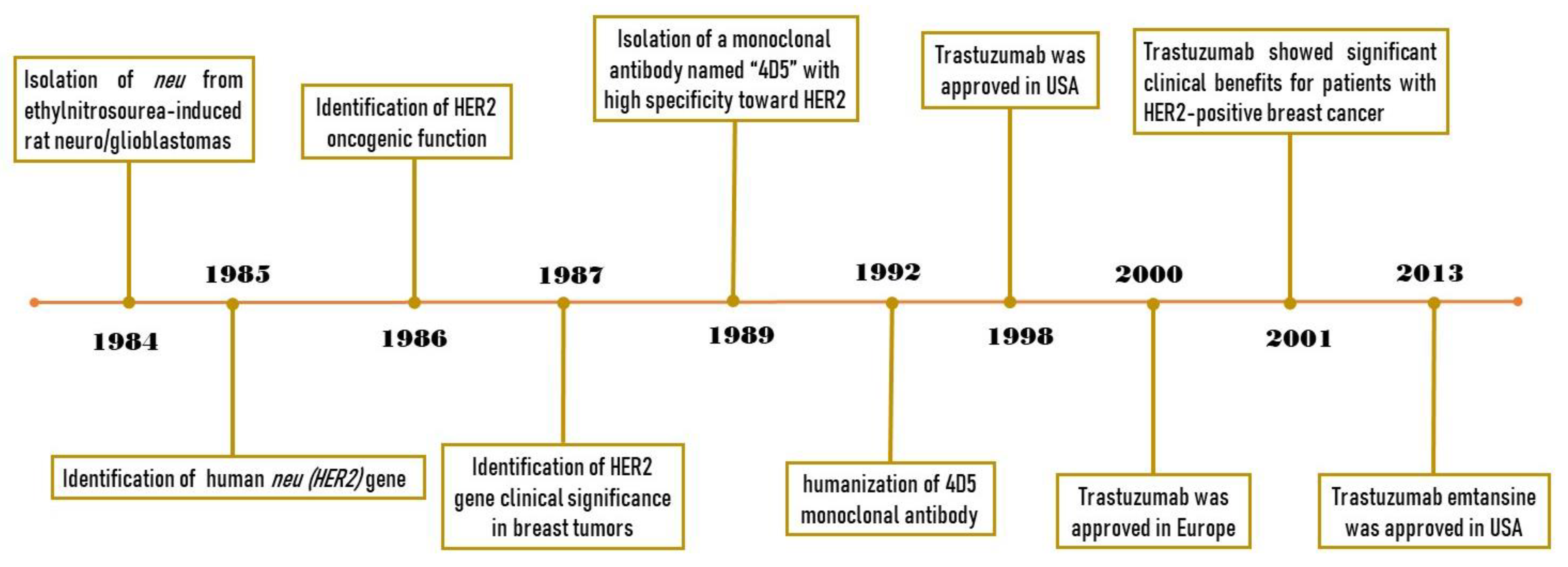
2. From Characterization of HER2 to Developing Trastuzumab
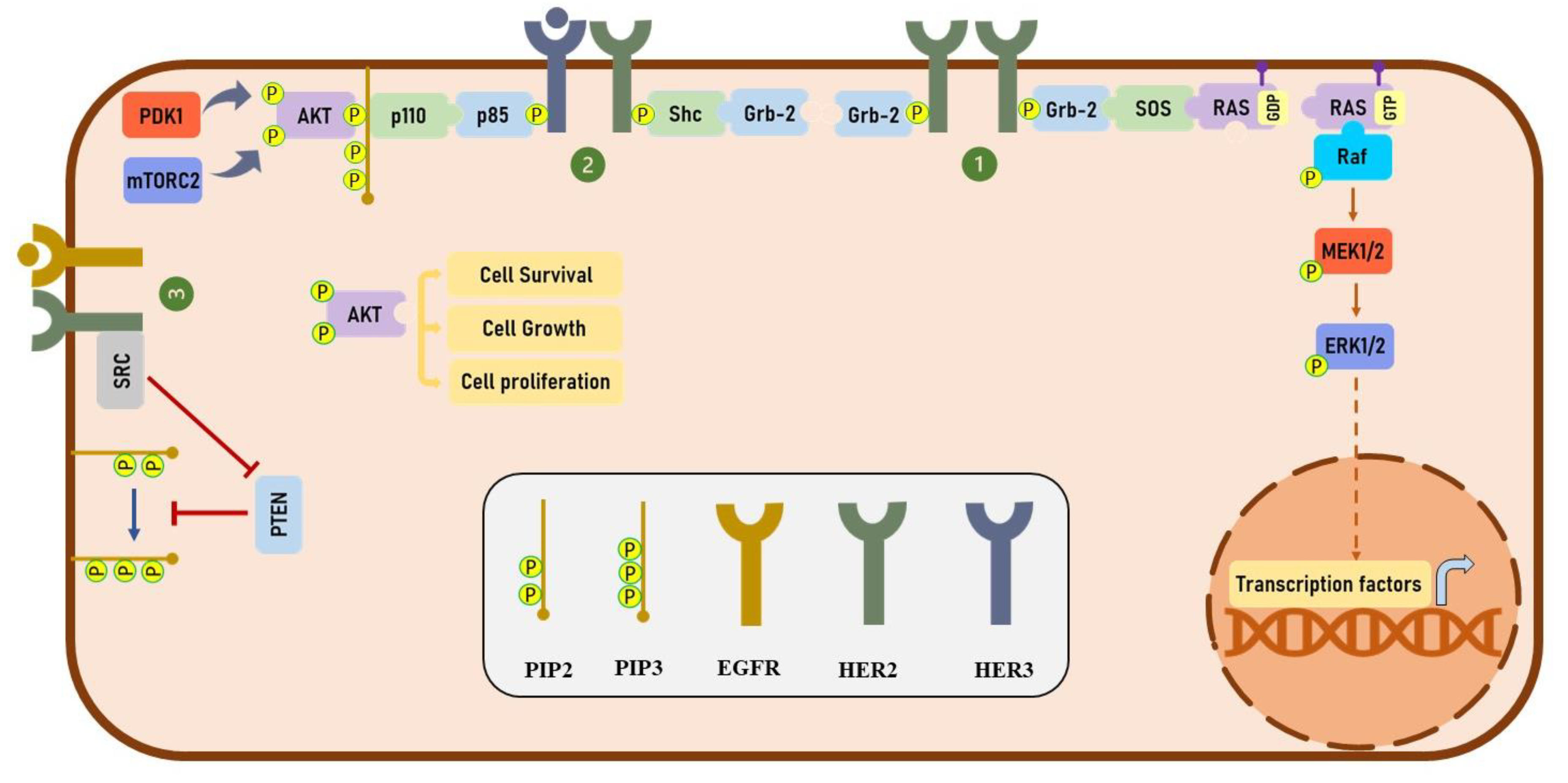
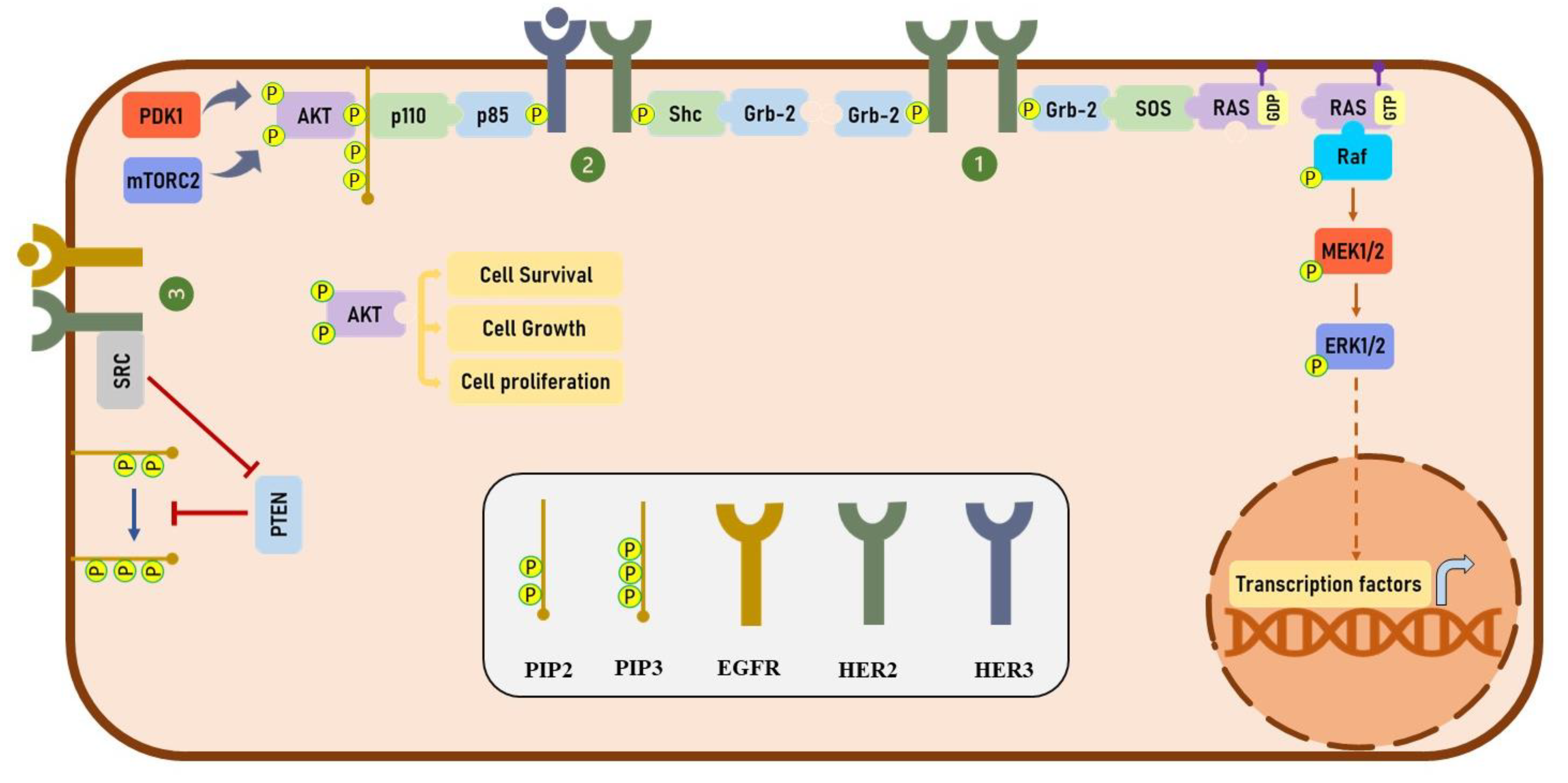
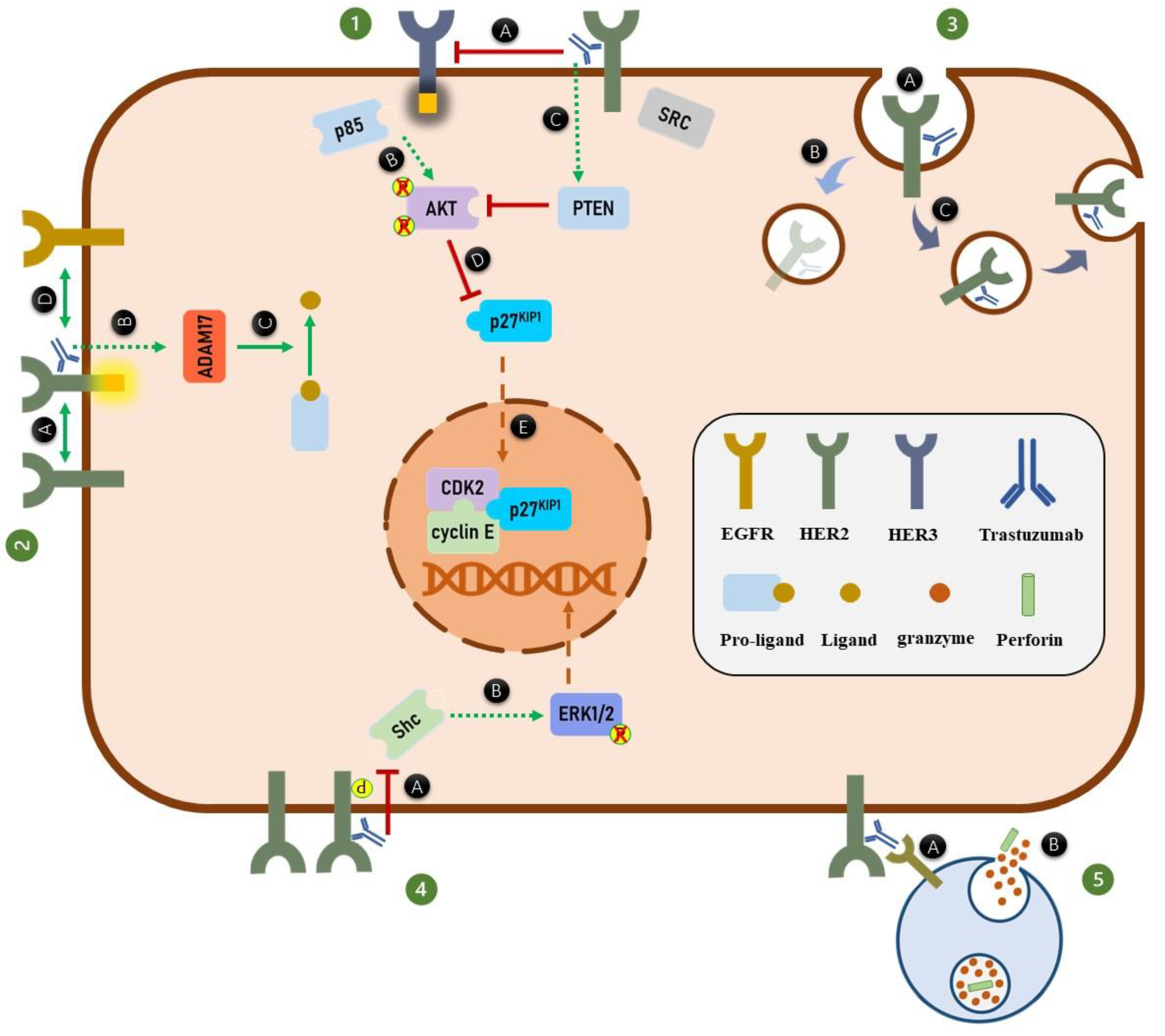
3. HER2 Signaling
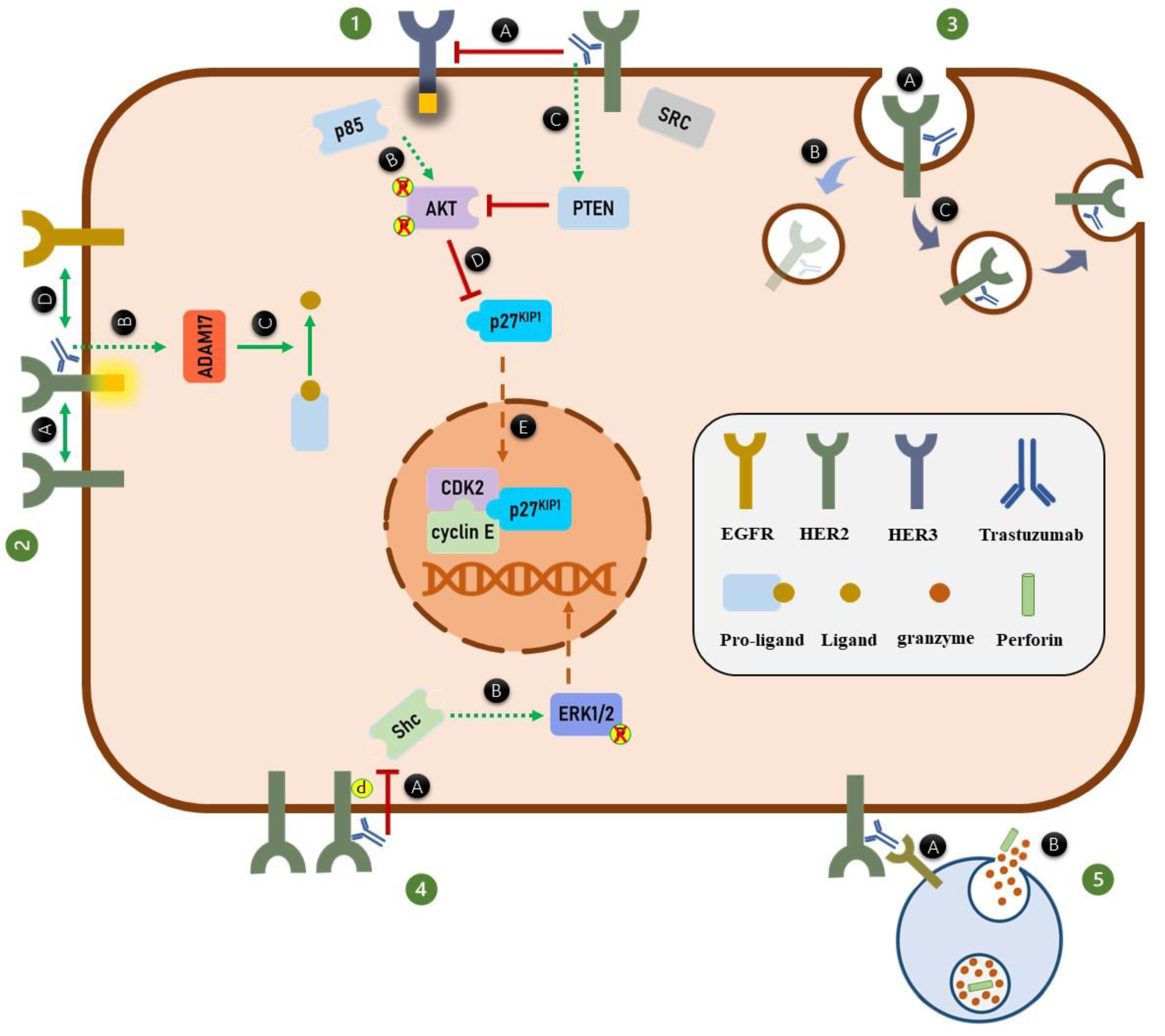
4. Trastuzumab and HER2 Dimerization
5. Trastuzumab and HER Phosphorylation
6. Trastuzumab and HER2 Endocytosis and Degradation
7. Trastuzumab and the MAPK Signaling Pathway
8. Trastuzumab and PI3K/AKT Signaling Pathway
9. Trastuzumab and Cell Cycle Arrest
10. Trastuzumab and Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC)
11. Trastuzumab and HER2 Isoforms
12. Mechanisms of Resistance to Trastuzumab
13. Conclusions and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Schechter, A.L.; Stern, D.F.; Vaidyanathan, L.; Decker, S.J.; Drebin, J.A.; Greene, M.I.; Weinberg, R.A. The neu oncogene: An erb-B-related gene encoding a 185,000-Mr tumour antigen. Nat. Cell Biol. 1984, 312, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.; Yang-Feng, T.L.; Liao, Y.C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, T.; Schlessinger, J.; Francke, U.; et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar] [CrossRef]
- Drebin, J.A.; Link, V.C.; Weinberg, R.A.; Greene, M.I. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proc. Natl. Acad. Sci. USA 1986, 83, 9129–9133. [Google Scholar] [CrossRef] [Green Version]
- Hudziak, R.M.; Schlessinger, J.; Ullrich, A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1987, 84, 7159–7163. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 1989, 9, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Sawyers, C.L. Herceptin: A First Assault on Oncogenes that Launched a Revolution. Cell 2019, 179, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Woelderink, A.; Ibarreta, D.; Hopkins, M.M.; Rodríguez-Cerezo, E. The current clinical practice of pharmacogenetic testing in Europe: TPMT and HER2 as case studies. Pharm. J. 2005, 6, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Yarden, Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A Structural Perspective on the Regulation of the Epidermal Growth Factor Receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Nami, B.; Maadi, H.; Wang, Z. Mechanisms Underlying the Action and Synergism of Trastuzumab and Pertuzumab in Targeting HER2-Positive Breast Cancer. Cancers 2018, 10, 342. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-S.; Mason, K.; Ramyar, K.X.; Stanley, A.M.; Gabelli, S.; Denney, D.W.; Leahy, D.J. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nat. Cell Biol. 2003, 421, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Diermeier, S.; Horváth, G.; Knuechel-Clarke, R.; Hofstaedter, F.; Szöllősi, J.; Brockhoff, G. Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp. Cell Res. 2005, 304, 604–619. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular Mechanisms of Epidermal Growth Factor Receptor (EGFR) Activation and Response to Gefitinib and Other EGFR-Targeting Drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Wallasch, C.; Weiss, F.U.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, N.; Cheng, K.; Koland, J. ErbB3 (HER3) interaction with the p85 regulatory subunit of phosphoinositide 3-kinase. Biochem. J. 1998, 333, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Lan, K.-H.; Zhou, X.; Tan, M.; Esteva, F.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M.M.; Gulizia, N.P. HER2 Amplification in Tumors Activates PI3K/Akt Signaling Independent of HER3. Cancer Res. 2018, 78, 3645–3658. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wang, L.; Shen, Y.; Wang, C.; Zhang, Y.; Meng, Y.; Yang, Y.; Liang, B.; Zhou, B.; Wang, H.; et al. Targeting EGFR/HER2 heterodimerization with a novel anti-HER2 domain II/III antibody. Mol. Immunol. 2017, 87, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Phillips, G.D.L.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-Independent HER2/HER3/PI3K Complex Is Disrupted by Trastuzumab and Is Effectively Inhibited by the PI3K Inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Gaborit, N.; Larbouret, C.; Vallaghe, J.; Peyrusson, F.; Bascoul-Mollevi, C.; Crapez, E.; Azria, D.; Chardès, T.; Poul, M.-A.; Mathis, G.; et al. Time-resolved Fluorescence Resonance Energy Transfer (TR-FRET) to Analyze the Disruption of EGFR/HER2 Dimers. J. Biol. Chem. 2011, 286, 11337–11345. [Google Scholar] [CrossRef] [Green Version]
- Maadi, H.; Nami, B.; Tong, J.; Li, G.; Wang, Z. The effects of trastuzumab on HER2-mediated cell signaling in CHO cells expressing human HER2. BMC Cancer 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colagar, A.H.; Amjadi, O.; Valadan, R.; Rafiei, A. Minimal HER1 and HER2 expressions in CHO and HEK-293 cells cause them appropriate negative cells for HERs-related studies. Res. Mol. Med. 2013, 1, 6–12. [Google Scholar] [CrossRef]
- Hatakeyama, M.; Yumoto, N.; Yu, X.; Shirouzu, M.; Yokoyama, S.; Konagaya, A. Transformation potency of ErbB heterodimer signaling is determined by B-Raf kinase. Oncogene 2004, 23, 5023–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokmanovic, M.; Wu, Y.; Shen, Y.; Chen, J.; Hirsch, D.S.; Wu, W.J. Trastuzumab-induced recruitment of Csk-homologous kinase (CHK) to ErbB2 receptor is associated with ErbB2-Y1248 phosphorylation and ErbB2 degradation to mediate cell growth inhibition. Cancer Biol. Ther. 2014, 15, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Diermeier-Daucher, S.; Breindl, S.; Buchholz, S.; Ortmann, O.; Brockhoff, G. Modular anti-EGFR and anti-Her2 targeting of SK-BR-3 and BT474 breast cancer cell lines in the presence of ErbB receptor-specific growth factors. Cytom. Part A 2011, 79, 684–693. [Google Scholar] [CrossRef]
- Bagnato, P.; Castagnino, A.; Cortese, K.; Bono, M.; Grasso, S.; Bellese, G.; Daniele, T.; Lundmark, R.; Defilippi, P.; Castagnola, P.; et al. Cooperative but distinct early co-signaling events originate from ERBB2 and ERBB1 receptors upon trastuzumab treatment in breast cancer cells. Oncotarget 2017, 8, 60109–60122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gijsen, M.; King, P.; Perera, T.; Parker, P.; Harris, A.; Larijani, B.; Kong, A. HER2 Phosphorylation Is Maintained by a PKB Negative Feedback Loop in Response to Anti-HER2 Herceptin in Breast Cancer. PLoS Biol. 2010, 8, e1000563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Hirsch, D.S.; Shen, Y.; Wu, W.J. Rac1 contributes to trastuzumab resistance of breast cancer cells: Rac1 as a potential therapeutic target for the treatment of trastuzumab-resistant breast cancer. Mol. Cancer Ther. 2009, 8, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Di Cara, G.; Marengo, G.; Albanese, N.N.; Marabeti, M.R.; Musso, R.; Cancemi, P.; Pucci-Minafra, I. Proteomic profiling of Trastuzumab (Herceptin(R))-sensitive and -resistant SKBR-3 breast cancer cells. Anticancer. Res. 2013, 33, 489–503. [Google Scholar]
- Milella, M.; Trisciuoglio, D.; Bruno, T.; Ciuffreda, L.; Mottolese, M.; Cianciulli, A.; Cognetti, F.; Zangemeister-Wittke, U.; Del Bufalo, D.; Zupi, G. Trastuzumab Down-Regulates Bcl-2 Expression and Potentiates Apoptosis Induction by Bcl-2/Bcl-XL Bispecific Antisense Oligonucleotides in HER-2Gene–Amplified Breast Cancer Cells. Clin. Cancer Res. 2004, 10, 7747–7756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Cajal, S.R.Y.; Arribas, J.; et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2008, 28, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Austin, C.D.; De Mazière, A.M.; Pisacane, P.I.; Van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and Sorting of ErbB2 and the Site of Action of Cancer Therapeutics Trastuzumab and Geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef]
- Mohsin, S.K.; Weiss, H.L.; Gutierrez, M.C.; Chamness, G.C.; Schiff, R.; DiGiovanna, M.P.; Wang, C.-X.; Hilsenbeck, S.G.; Osborne, C.K.; Allred, D.C.; et al. Neoadjuvant Trastuzumab Induces Apoptosis in Primary Breast Cancers. J. Clin. Oncol. 2005, 23, 2460–2468. [Google Scholar] [CrossRef]
- Pereira, P.; Sharma, S.K.; Carter, L.; Edwards, K.J.; Pourat, J.; Ragupathi, A.; Janjigian, Y.Y.; Durack, J.C.; Lewis, J.S. Caveolin-1 mediates cellular distribution of HER2 and affects trastuzumab binding and therapeutic efficacy. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Korzeniecki, C.; Priefer, R. Targeting KRAS mutant cancers by preventing signaling transduction in the MAPK pathway. Eur. J. Med. Chem. 2021, 211, 113006. [Google Scholar] [CrossRef]
- Soares-Silva, M.C.; Diniz, F.F.; Gomes, G.N.; Ebahia, D. The Mitogen-Activated Protein Kinase (MAPK) Pathway: Role in Immune Evasion by Trypanosomatids. Front. Microbiol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.-D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nat. Cell Biol. 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta (BBA)-Bioenergy 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Jin, X.; Lin, J. Stat3 upregulates MEK5 expression in human breast cancer cells. Oncogene 2004, 23, 8301–8309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaoka, Y.; Nishida, K.; Fujio, Y.; Izumi, M.; Terai, K.; Oshima, Y.; Sugiyama, S.; Matsuda, S.; Koyasu, S.; Yamauchi-Takihara, K.; et al. Activation of gp130 transduces hypertrophic signal through interaction of scaffolding/docking protein Gab1 with tyrosine phosphatase SHP2 in cardiomyocytes. Circ. Res. 2003, 93, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; Chinratanalab, W.; Ritter, C.A.; King, W.; Seelig, S.; Arteaga, C.L. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt Is required for antibody-mediated effects on p27, cyclin D1, and antitumor action. Cancer Res. 2002, 62, 4132–4141. [Google Scholar] [PubMed]
- Wang, C.; Wang, L.; Yu, X.; Zhang, Y.; Meng, Y.; Wang, H.; Yang, Y.; Gao, J.; Wei, H.; Zhao, J.; et al. Combating acquired resistance to trastuzumab by an anti-ErbB2 fully human antibody. Oncotarget 2017, 8, 42742–42751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Narasanna, A.; Wang, S.E.; Liu, S.; Chakrabarty, A.; Balko, J.M.; González-Angulo, A.M.; Mills, G.B.; Penuel, E.; Winslow, J.; et al. Trastuzumab Has Preferential Activity against Breast Cancers Driven by HER2 Homodimers. Cancer Res. 2011, 71, 1871–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, A.H.; Zweemer, A.J.; Nagumo, Y.; Faratian, D.; Muir, M.; Dodds, M.; Um, I.; Kay, C.; Hasmann, M.; Harrison, D.J.; et al. Defining the molecular response to trastuzumab, pertuzumab and combination therapy in ovarian cancer. Br. J. Cancer 2012, 106, 1779–1789. [Google Scholar] [CrossRef]
- Dubska, L.; Andera, L.; Sheard, M.A. HER2 signaling downregulation by trastuzumab and suppression of the PI3K/Akt pathway: An unexpected effect on TRAIL-induced apoptosis. FEBS Lett. 2005, 579, 4149–4158. [Google Scholar] [CrossRef] [Green Version]
- Delord, J.-P.; Quideau, S.; Rochaix, P.; Caselles, O.; Couderc, B.; Hennebelle, I.; Courbon, F.; Canal, P.; Allal, B.C. Trastuzumab induced in vivo tissue remodelling associated in vitro with inhibition of the active forms of AKT and PTEN and RhoB induction in an ovarian carcinoma model. Br. J. Cancer 2010, 103, 61–72. [Google Scholar] [CrossRef]
- Watanabe, S.; Yonesaka, K.; Tanizaki, J.; Nonagase, Y.; Takegawa, N.; Haratani, K.; Kawakami, H.; Hayashi, H.; Takeda, M.; Tsurutani, J.; et al. Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Med. 2019, 8, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Carmona, F.J.; Montemurro, F.; Kannan, S.; Rossi, V.; Verma, C.; Baselga, J.; Scaltriti, M. AKT signaling in ERBB2-amplified breast cancer. Pharmacol. Ther. 2016, 158, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Vara, J.; Ángel, F.; Casado, E.; De Castro, J.; Cejas, P.; Iniesta, C.B.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Soltoff, S.P.; Carraway, K.L.; Prigent, S.A.; Gullick, W.G.; Cantley, L.C. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol. Cell. Biol. 1994, 14, 3550–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkas-Kramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M.; et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cron, P.; Good, V.M.; Thompson, V.; Hemmings, B.A.; Barford, D. Crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Genet. 2002, 9, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Ali-Hemmati, A.; Rahmati, M.; Charoudeh, H.N. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Le, X.-F.; Lammayot, A.; Gold, D.; Lu, Y.; Mao, W.; Chang, T.; Patel, A.; Mills, G.B.; Bast, R. Genes Affecting the Cell Cycle, Growth, Maintenance, and Drug Sensitivity Are Preferentially Regulated by Anti-HER2 Antibody through Phosphatidylinositol 3-Kinase-AKT Signaling. J. Biol. Chem. 2005, 280, 2092–2104. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.A.; Siminovitch, K.A.; et al. Src Family Protein-tyrosine Kinases Alter the Function of PTEN to Regulate Phosphatidylinositol 3-Kinase/AKT Cascades. J. Biol. Chem. 2003, 278, 40057–40066. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, T.O.P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Sherr, C.J. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef] [Green Version]
- Trimarchi, J.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef]
- Wander, S.A.; Zhao, D.; Slingerland, J.M. p27: A Barometer of Signaling Deregulation and Potential Predictor of Response to Targeted Therapies. Clin. Cancer Res. 2011, 17, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlach, J.; Hennecke, S.; Amati, B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J. 1997, 16, 5334–5344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, N.; Kitagawa, M.; Hatakeyama, S.; Nakayama, K.-I. Phosphorylation at Serine 10, a Major Phosphorylation Site of p27, Increases Its Protein Stability. J. Biol. Chem. 2000, 275, 25146–25154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, X.-F.; Claret, F.-X.; Lammayot, A.; Tian, L.; Deshpande, D.; LaPushin, R.; Tari, A.M.; Bast, R.C. The Role of Cyclin-dependent Kinase Inhibitor p27Kip1 in Anti-HER2 Antibody-induced G1 Cell Cycle Arrest and Tumor Growth Inhibition. J. Biol. Chem. 2003, 278, 23441–23450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motti, M.L.; De Marco, C.; Califano, D.; Fusco, A.; Viglietto, G. Akt-dependent T198 phosphorylation of cyclin-dependent kinase inhibitor p27kip1 in breast cancer. Cell Cycle 2004, 3, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Larrea, M.D.; Wander, S.A.; Slingerland, J. p27 as Jekyll and Hyde: Regulation of cell cycle and cell motility. Cell Cycle 2009, 8, 3455–3461. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Song, J.; Park, S.; Ham, S.; Paek, K.; Kang, M.; Chae, Y.; Seo, H.; Kim, H.-C.; Flores, M. Drifts in ADCC-related quality attributes of Herceptin®: Impact on development of a trastuzumab biosimilar. MAbs 2017, 9, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Wei, F.; Wang, L.; Yu, W.; Zhang, N.; Zhang, X.; Han, Y.; Yu, J.; Ren, X. Herceptin Enhances the Antitumor Effect of Natural Killer Cells on Breast Cancer Cells Expressing Human Epidermal Growth Factor Receptor-2. Front. Immunol. 2017, 8, 1426. [Google Scholar] [CrossRef] [Green Version]
- De Maria, A.; Bozzano, F.; Cantoni, C.; Moretta, L. Revisiting human natural killer cell subset function revealed cytolytic CD56dimCD16+ NK cells as rapid producers of abundant IFN- on activation. Proc. Natl. Acad. Sci. USA 2011, 108, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Kute, T.; Stehle, J.J.R.; Ornelles, D.; Walker, N.; Delbono, O.; Vaughn, J.P. Understanding key assay parameters that affect measurements of trastuzumab-mediated ADCC against Her2 positive breast cancer cells. Oncoimmunology 2012, 1, 810–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treffers, L.W.; Van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; Van Der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.J.J.H.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcγRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front. Immunol. 2019, 9, 3124. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortum, R.L.; Rouquette-Jazdanian, A.K.; Samelson, L.E. Ras and extracellular signal-regulated kinase signaling in thymocytes and T cells. Trends Immunol. 2013, 34, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, D.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. Differential Regulation of Human NK Cell-Mediated Cytotoxicity by the Tyrosine Kinase Itk. J. Immunol. 2007, 178, 3575–3582. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-G.; Liu, Y.; Chen, F. Soluble fibrinogen like protein 2 (sFGL2), the novel effector molecule for immunoregulation. Oncotarget 2016, 8, 3711–3723. [Google Scholar] [CrossRef] [Green Version]
- Gumbleton, M.; Kerr, W.G. Role of inositol phospholipid signaling in natural killer cell biology. Front. Immunol. 2013, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, J. Anatomy of a murder: How cytotoxic T cells and NK cells are activated, develop, and eliminate their targets. Immunol. Rev. 2010, 235, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Prager, I.; Liesche, C.; Van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandström, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor–mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [Green Version]
- Rainone, V.; Martelli, C.; Ottobrini, L.; Biasin, M.; Borelli, M.; Lucignani, G.; Trabattoni, D.; Clerici, M. Immunological Characterization of Whole Tumour Lysate-Loaded Dendritic Cells for Cancer Immunotherapy. PLoS ONE 2016, 11, e0146622. [Google Scholar] [CrossRef] [PubMed]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef] [Green Version]
- Collins, D.M.; O’Donovan, N.; McGowan, P.; O’Sullivan, F.; Duffy, M.J.; Crown, J. Trastuzumab induces antibody-dependent cell-mediated cytotoxicity (ADCC) in HER-2-non-amplified breast cancer cell lines. Ann. Oncol. 2012, 23, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J. Transl. Med. 2013, 11, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, H.Y.; De Lima, P.O.; Cruz, J.L.G.; Banushi, B.; Echejoh, G.; Hu, L.; Joseph, S.R.; Lum, B.; Rae, J.; O’Donnell, J.S.; et al. Endocytosis Inhibition in Humans to Improve Responses to ADCC-Mediating Antibodies. Cell 2020, 180, 895–914.e27. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kono, K.; Mizukami, Y.; Mimura, K.; Fujii, H. Mechanisms of escape from trastuzumab-mediated ADCC in esophageal squamous cell carcinoma: Relation to susceptibility to perforin-granzyme. Anticancer. Res. 2009, 29, 2137–2146. [Google Scholar]
- Boero, S.; Morabito, A.; Banelli, B.; Cardinali, B.; Dozin, B.; Lunardi, G.; Piccioli, P.; Lastraioli, S.; Carosio, R.; Salvi, S.; et al. Analysis of in vitro ADCC and clinical response to trastuzumab: Possible relevance of FcγRIIIA/FcγRIIA gene polymorphisms and HER-2 expression levels on breast cancer cell lines. J. Transl. Med. 2015, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Woof, J.M.; Hart, S.P.; Dransfield, I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin. Exp. Immunol. 2009, 157, 244–254. [Google Scholar] [CrossRef]
- Bang, Y.J.; Giaccone, G.; Im, S.A.; Oh, D.Y.; Bauer, T.M.; Nordstrom, J.L.; Li, H.; Chichili, G.R.; Moore, P.A.; Hong, S.; et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017, 28, 855–861. [Google Scholar] [CrossRef]
- Tural, D.; Akar, E.; Mutlu, H.; Kilickap, S. P95 HER2 fragments and breast cancer outcome. Expert Rev. Anticancer Ther. 2014, 14, 1089–1096. [Google Scholar] [CrossRef]
- Anido, J.; Scaltriti, M.; Bech-Serra, J.J.; Josefat, B.S.; Todo, F.R.; Baselga, J.; Arribas, J. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J. 2006, 25, 3234–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumsri, S.; Sperinde, J.; Liu, H.; Gligorov, J.; Spano, J.-P.; Antoine, M.; Aspitia, A.M.; Tan, W.; Winslow, J.; Petropoulos, C.J.; et al. High p95HER2/HER2 Ratio Associated With Poor Outcome in Trastuzumab-Treated HER2-Positive Metastatic Breast Cancer NCCTG N0337 and NCCTG 98-32-52 (Alliance). Clin. Cancer Res. 2018, 24, 3053–3058. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Cajal, S.R.Y.; Arribas, J.; et al. Expression of p95HER2, a Truncated Form of the HER2 Receptor, and Response to Anti-HER2 Therapies in Breast Cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, K.; Angelini, P.-D.; Laos, S.; Bach-Faig, A.; Cunningham, M.P.; Ferrer-Ramón, C.; Luque-García, A.; Garcia-Castillo, J.; Parra-Palau, J.L.; Scaltriti, M.; et al. A Naturally Occurring HER2 Carboxy-Terminal Fragment Promotes Mammary Tumor Growth and Metastasis. Mol. Cell. Biol. 2009, 29, 3319–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and Breast Cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, M.A.; Codony-Servat, J.; Albanell, J.; Rojo, F.; Arribas, J.; Baselga, J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61, 4744–4749. [Google Scholar]
- Parra-Palau, J.L.; Morancho, B.; Peg, V.; Escorihuela, M.; Scaltriti, M.; Vicario, R.; Zacarias-Fluck, M.; Pedersen, K.; Pandiella, A.; Nuciforo, P.; et al. Effect of p95HER2/611CTF on the Response to Trastuzumab and Chemotherapy. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [Green Version]
- Zagouri, F.; Bournakis, E.; Koutsoukos, K.; Papadimitriou, C.A. Heat Shock Protein 90 (Hsp90) Expression and Breast Cancer. Pharmaceuticals 2012, 5, 1008–1020. [Google Scholar] [CrossRef] [Green Version]
- Crouch, B.; Murphy, H.; Belonwu, S.; Martinez, A.; Gallagher, J.; Hall, A.; Soo, M.S.; Lee, M.; Hughes, P.; Haystead, T.; et al. Leveraging ectopic Hsp90 expression to assay the presence of tumor cells and aggressive tumor phenotypes in breast specimens. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Scaltriti, M.; Angelini, P.; Ye, Q.; Guzman, M.; Hudis, C.A.; Norton, L.; Solit, D.B.; Arribas, J.; Baselga, J.; et al. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene 2009, 29, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.; Balsari, A.; Ménard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, L.; Iezzi, M.; Ghedini, G.C.; Ciravolo, V.; Marzano, G.; Lamolinara, A.; Zappasodi, R.; Gasparini, P.; Campiglio, M.; Amici, A.; et al. Activated d16HER2 Homodimers and SRC Kinase Mediate Optimal Efficacy for Trastuzumab. Cancer Res. 2014, 74, 6248–6259. [Google Scholar] [CrossRef] [Green Version]
- Palladini, A.; Nicoletti, G.; Lamolinara, A.; Dall’Ora, M.; Balboni, T.; Ianzano, M.L.; Laranga, R.; Landuzzi, L.; Giusti, V.; Ceccarelli, C.; et al. HER2 isoforms co-expression differently tunes mammary tumor phenotypes affecting onset, vasculature and therapeutic response. Oncotarget 2017, 8, 54444–54458. [Google Scholar] [CrossRef]
- Bartsch, R.; Wenzel, C.; Steger, G.G. Trastuzumab in the management of early and advanced stage breast cancer. Biol. Targets Ther. 2007, 1, 19–31. [Google Scholar]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A Functional Genetic Approach Identifies the PI3K Pathway as a Major Determinant of Trastuzumab Resistance in Breast Cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, G.K.; Robles, R.; Park, J.W.; Montgomery, P.A.; Daniel, J.; Holmes, W.E.; Lee, J.; Keller, G.A.; Li, W.L.; Fendly, B.M. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Mol. Cell. Biol. 1993, 13, 2247–2257. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-Like Growth Factor-I Receptor Signaling and Resistance to Trastuzumab (Herceptin). J. Natl. Cancer Inst. 2001, 93, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Peddi, P.F.; Hurvitz, S.A. Ado-trastuzumab emtansine (T-DM1) in human epidermal growth factor receptor 2 (HER2)-positive metastatic breast cancer: Latest evidence and clinical potential. Ther. Adv. Med Oncol. 2014, 6, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Roman, L.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Staroslawska, E.; Rodríguez, J.R.D.L.H.; Im, S.-A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef]
- Johnston, S.R.; Hegg, R.; Im, S.-A.; Park, I.H.; Burdaeva, O.; Kurteva, G.; Press, M.F.; Tjulandin, S.; Iwata, H.; Simon, S.D.; et al. Phase III, Randomized Study of Dual Human Epidermal Growth Factor Receptor 2 (HER2) Blockade With Lapatinib Plus Trastuzumab in Combination With an Aromatase Inhibitor in Postmenopausal Women With HER2-Positive, Hormone Receptor–Positive Metastatic Breast Cancer: ALTERNATIVE. J. Clin. Oncol. 2018, 36, 741–748. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Mechanism of Action | Finding | Experimental Model | Method | Reference |
|---|---|---|---|---|
| Effect of trastuzumab on HER2 homodimerization | Activatory effect | CHO cells transfected with HER2 receptors | Cross-linking assay | [26] |
| BT474 and SKBR3 HER2-positive BC cells | FRET * | [16] | ||
| Effect of trastuzumab on ligand-dependent HER2 heterodimerization | No effect | BT474 and SKBR3 HER2-positive BC cells | Co-IP ** | [23] |
| No effect | SKBR3 HER2-positive BC cells | Co-IP | [24] | |
| Effect of trastuzumab on ligand-independent HER2 heterodimerization | Inhibitory effect | SKBR3 HER2-positive BC cells | Reversible cross-linking followed by Co-IP | [24] |
| Inhibitory effect | SKOV3 HER2-positive ovarian cancer cells | TR-FRET *** | [25] | |
| No effect | BT474 and SKBR3 HER2-positive BC cells | FRET | [16] | |
| Effect of trastuzumab on HER2 phosphorylation | Activatory effect | SKBR3 and BT474 HER2-positive BC cells | WB **** | [29,31,32] |
| Effect of trastuzumab on HER3 phosphorylation | Inhibitory effect | SKBR3 and BT474 HER2-positive BC cells | WB | [24,29] |
| Effect of trastuzumab on HER2 endocytosis and downregulation | Activatory effect | SKBR3 HER2-positive BC cells | ICC # and WB | [34] |
| Activatory effect | HER2-positive BC tumor samples | IHC ## | [36] | |
| No effect | SKBR3 HER2-positive BC cells | ICC and WB | [31,38] | |
| Effect of trastuzumab on MAPK signaling pathway | Inhibitory effect | SKBR3 and BT474 HER2-positive BC cells | WB | [52] |
| Inhibitory effect | NCI-N87 HER2-positive gastric cell line | WB | [53] | |
| Inhibitory effect | MCF10A cells transfected with chimeric HER2 and FK506-binding protein (FKBP) | WB | [54] | |
| Activatory effect | SKBR3 and BT474 HER2-positive BC cells | WB | [29,31] | |
| Effect of trastuzumab on PI3K/AKT signaling pathway | Inhibitory effect | SKBR3 HER2-positive BC cells | ELISA ### | [24] |
| Inhibitory effect | SKBR3 and BT474 HER2-positive BC cells | WB | [29,52,67] | |
| Effect of trastuzumab on cell cycle | Inhibitory effect | SKBR3 and BT474 HER2-positive BC cells | DNA content quantification using flow cytometry | [30,76] |
| Effect of trastuzumab on ADCC | Activatory effect | CHO cells transfected with HER2 receptors | Promega ADCC Bioassay kit | [26] |
| Activatory effect | SKBR3 HER2-positive BC cells | live-cell imaging | [80] | |
| Effect of trastuzumab on HER2 cleavage | Inhibitory effect | SKBR3 and BT474 HER2-positive BC cells | WB | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maadi, H.; Soheilifar, M.H.; Choi, W.-S.; Moshtaghian, A.; Wang, Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers 2021, 13, 3540. https://doi.org/10.3390/cancers13143540
Maadi H, Soheilifar MH, Choi W-S, Moshtaghian A, Wang Z. Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers. 2021; 13(14):3540. https://doi.org/10.3390/cancers13143540
Chicago/Turabian StyleMaadi, Hamid, Mohammad Hasan Soheilifar, Won-Shik Choi, Abdolvahab Moshtaghian, and Zhixiang Wang. 2021. "Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma" Cancers 13, no. 14: 3540. https://doi.org/10.3390/cancers13143540
APA StyleMaadi, H., Soheilifar, M. H., Choi, W.-S., Moshtaghian, A., & Wang, Z. (2021). Trastuzumab Mechanism of Action; 20 Years of Research to Unravel a Dilemma. Cancers, 13(14), 3540. https://doi.org/10.3390/cancers13143540