
Targeting Metastatic Colorectal Cancer with Immune Oncological Therapies

Abstract
:Simple Summary
Abstract
1. Introduction
2. Colorectal Cancer—Personalised Medicine for a Heterogeneous Disease
2.1. Classification by Mutational Status of CRC
2.2. Role of Immunotherapy in Serrated Adenocarcinoma of the Colon
2.3. Consensus Molecular Subtypes in CRC
2.4. Transcriptomic Profiling of Metastatic Colorectal Cancer
3. Colorectal Cancer and the Tumour Microenvironment—An Immunological Basis for Targeted Therapy
3.1. T-Cells
3.2. Natural Killer Cells
3.3. Dendritic Cells
3.4. Myeloid Cells
3.5. Neutrophils
3.6. Translating Immunopathology Understanding into Therapeutics
4. MMR Deficient—MSI-High Metastatic Colorectal Cancer
5. MSS Metastatic Colorectal Cancer
6. Combination Strategies in Metastatic Colorectal Cancer—Overcoming Resistance
6.1. Immunotherapy and MEK Inhibitors
6.2. Immunotherapy and CEA Inhibition
6.3. Immunotherapy and IDO Inhibitors
6.4. Immunotherapy and Radiotherapy
7. Preclinical Studies Investigating Promising Immunotherapy Strategies in Metastatic Colorectal Cancer
8. Genomic Biomarkers of Response to Immunotherapy
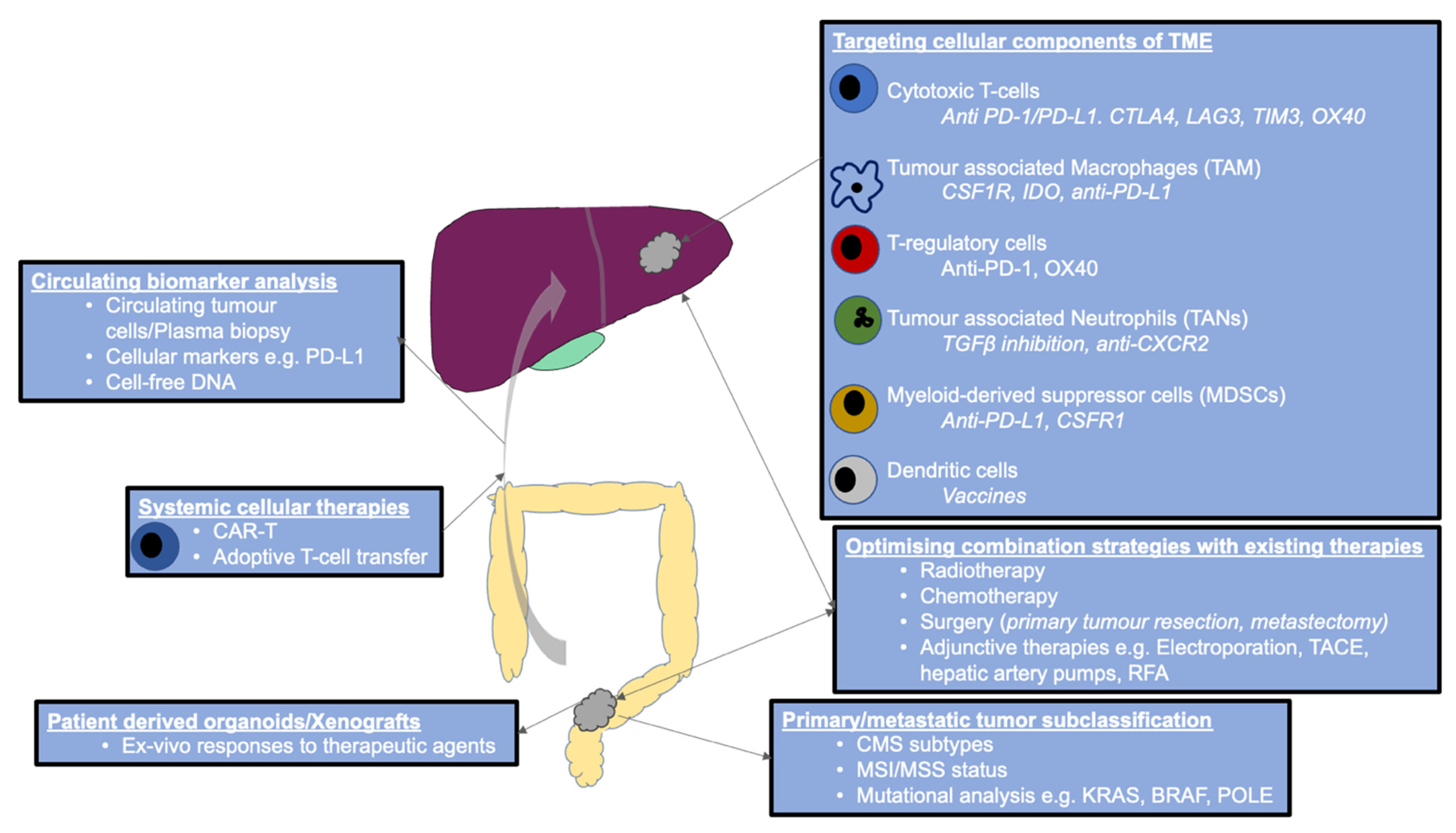
9. Strategies to Improve Outcomes
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Rivera, F.; Karthaus, M.; Fasola, G.; Canon, J.L.; Hecht, J.R.; Yu, H.; Oliner, K.S.; Go, W.Y. PEAK: A randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J. Clin. Oncol. 2014, 32, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Ye, J.; Lin, M.; Zhang, C.; Zhu, X.; Li, S.; Liu, H.; Yin, J.; Yu, H.; Zhu, K. Tissue gene mutation profiles in patients with colorectal cancer and their clinical implications. Biomed. Rep. 2020, 13, 43–48. [Google Scholar] [CrossRef]
- Leslie, A.; Carey, F.A.; Pratt, N.R.; Steele, R.J. The colorectal adenoma-carcinoma sequence. Br. J. Surg. 2002, 89, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.B.; Venook, A.P.; Al-Hawary, M.M.; Arain, M.A.; Chen, Y.J.; Ciombor, K.K.; Cohen, S.; Cooper, H.S.; Deming, D.; Farkas, L.; et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 329–359. [Google Scholar] [CrossRef]
- Gau, L.; Ribeiro, M.; Pereira, B.; Poirot, K.; Dupré, A.; Pezet, D.; Gagnière, J. Impact of BRAF mutations on clinical outcomes following liver surgery for colorectal liver metastases: An updated meta-analysis. Eur. J. Surg. Oncol. 2021, in press. [Google Scholar] [CrossRef]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Martens, U.M.; Riera-Knorrenschild, J.; Greeve, J.; Florschütz, A.; Wessendorf, S.; Ettrich, T.; Kanzler, S.; Nörenberg, D.; Ricke, J.; et al. FOLFOXIRI Plus Panitumumab As First-Line Treatment of RAS Wild-Type Metastatic Colorectal Cancer: The Randomized, Open-Label, Phase II VOLFI Study (AIO KRK0109). J. Clin. Oncol. 2019, 37, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- Boland, P.M.; Ma, W.W. Immunotherapy for Colorectal Cancer. Cancers 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Ottaiano, A.; Normanno, N.; Facchini, S.; Cassata, A.; Nappi, A.; Romano, C.; Silvestro, L.; De Stefano, A.; Rachiglio, A.M.; Roma, C.; et al. Study of Ras Mutations’ Prognostic Value in Metastatic Colorectal Cancer: STORIA Analysis. Cancers 2020, 12, 1919. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, S.; Matrone, N.; Muddassir, A.L.; Martini, G.; Sorokin, A.; De Falco, V.; Giunta, E.F.; Ciardiello, D.; Martinelli, E.; Belli, V.; et al. Triple blockade of EGFR, MEK and PD-L1 has antitumor activity in colorectal cancer models with constitutive activation of MAPK signaling and PD-L1 overexpression. J. Exp. Clin. Cancer Res. 2019, 38, 492. [Google Scholar] [CrossRef] [Green Version]
- Boland, P.M.; Hutsons, A.; Maguire, O.; Minderman, H.; Fountzilas, C.; Iyer, R.V. A phase Ib/II study of cetuximab and pembrolizumab in RAS-wt mCRC. J. Clin. Oncol. 2018, 36, 834. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Alexandrov, L.B. Unraveling the genomic landscape of colorectal cancer through mutational signatures. Adv. Cancer Res. 2021, 151, 385–424. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jass, J.R. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007, 50, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Goel, A.; Chung, D.C. Pathways of Colorectal Carcinogenesis. Gastroenterology 2020, 158, 291–302. [Google Scholar] [CrossRef] [PubMed]
- De Palma, F.D.E.; D’Argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Pitroda, S.P.; Khodarev, N.N.; Huang, L.; Uppal, A.; Wightman, S.C.; Ganai, S.; Joseph, N.; Pitt, J.; Brown, M.; Forde, M.; et al. Integrated molecular subtyping defines a curable oligometastatic state in colorectal liver metastasis. Nat. Commun. 2018, 9, 1793. [Google Scholar] [CrossRef]
- Shiomi, A.; Kusuhara, M.; Sugino, T.; Sugiura, T.; Ohshima, K.; Nagashima, T.; Urakami, K.; Serizawa, M.; Saya, H.; Yamaguchi, K. Comprehensive genomic analysis contrasting primary colorectal cancer and matched liver metastases. Oncol. Lett. 2021, 21, 466. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pagès, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Donadon, M.; Hudspeth, K.; Cimino, M.; Di Tommaso, L.; Preti, M.; Tentorio, P.; Roncalli, M.; Mavilio, D.; Torzilli, G. Increased Infiltration of Natural Killer and T Cells in Colorectal Liver Metastases Improves Patient Overall Survival. J. Gastrointest. Surg. 2017, 21, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, P.; Ahmed, S. Current status of immunotherapy in metastatic colorectal cancer. Int. J. Colorectal. Dis. 2019, 34, 13–25. [Google Scholar] [CrossRef]
- Galbraith, N.; Walker, S.; Galandiuk, S.; Gardner, S.; Polk, H.C., Jr. The Significance and Challenges of Monocyte Impairment: For the Ill Patient and the Surgeon. Surg. Infect. 2016, 17, 303–312. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602.e510. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, N.J.; Walker, S.P.; Gardner, S.A.; Bishop, C.; Galandiuk, S.; Polk, H.C., Jr. Interferon-gamma increases monocyte PD-L1 but does not diminish T-cell activation. Cell Immunol. 2020, 357, 104197. [Google Scholar] [CrossRef]
- Inagaki, K.; Kunisho, S.; Takigawa, H.; Yuge, R.; Oka, S.; Tanaka, S.; Shimamoto, F.; Chayama, K.; Kitadai, Y. Role of tumor-associated macrophages at the invasive front in human colorectal cancer progression. Cancer Sci. 2021, 112, 2692–2704. [Google Scholar] [CrossRef]
- Antoniotti, C.; Borelli, B.; Rossini, D.; Pietrantonio, F.; Morano, F.; Salvatore, L.; Lonardi, S.; Marmorino, F.; Tamberi, S.; Corallo, S.; et al. AtezoTRIBE: A randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer 2020, 20, 683. [Google Scholar] [CrossRef]
- Chen, E.X.; Jonker, D.J.; Loree, J.M.; Kennecke, H.F.; Berry, S.R.; Couture, F.; Ahmad, C.E.; Goffin, J.R.; Kavan, P.; Harb, M.; et al. Effect of Combined Immune Checkpoint Inhibition vs. Best Supportive Care Alone in Patients With Advanced Colorectal Cancer: The Canadian Cancer Trials Group CO.26 Study. JAMA Oncol. 2020, 6, 831–838. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair—Deficient/Microsatellite Instability—High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Overman, M.J.; Hartman, L.; Khoukaz, T.; Brutcher, E.; Lenz, H.J.; Atasoy, A.; Shangguan, T.; Zhao, H.; El-Rayes, B. Safety of Nivolumab plus Low-Dose Ipilimumab in Previously Treated Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer. Oncologist 2019, 24, 1453–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Kopetz, S.; McDermott, R.S.; Leach, J.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.D.; et al. Nivolumab ± ipilimumab in treatment (tx) of patients (pts) with metastatic colorectal cancer (mCRC) with and without high microsatellite instability (MSI-H): CheckMate-142 interim results. J. Clin. Oncol. 2016, 34, 3501. [Google Scholar] [CrossRef]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II Study of the Anti-Cytotoxic T-Lymphocyte–Associated Antigen 4 Monoclonal Antibody, Tremelimumab, in Patients With Refractory Metastatic Colorectal Cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R.; Falchook, G.S.; Hamada, K.; Makris, L.; Bendell, J.C. A phase 2 trial of trifluridine/tipiracil plus nivolumab in patients with heavily pretreated microsatellite-stable metastatic colorectal cancer. Cancer Med. 2021, 10, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Mettu, N.B.; Twohy, E.; Ou, F.-S.; Halfdanarson, T.; Lenz, H.; Breakstone, R.; Boland, P.; Crysler, O.; Wu, C.; Grothey, A.; et al. BACCI: A phase II randomized, double-blind, multicenter, placebo-controlled study of capecitabine (C) bevacizumab (B) plus atezolizumab (A) or placebo (P) in refractory metastatic colorectal cancer (mCRC): An ACCRU network study. Ann. Oncol. 2019, 30, v203. [Google Scholar] [CrossRef]
- Segal, N.H.; Cercek, A.; Ku, G.; Wu, A.J.; Rimner, A.; Khalil, D.N.; Reidy-Lagunes, D.; Cuaron, J.; Yang, T.J.; Weiser, M.R.; et al. Phase II Single-arm Study of Durvalumab and Tremelimumab with Concurrent Radiotherapy in Patients with Mismatch Repair-proficient Metastatic Colorectal Cancer. Clin. Cancer Res. 2021, 27, 2200–2208. [Google Scholar] [CrossRef]
- Li, J.; Cong, L.; Liu, J.; Peng, L.; Wang, J.; Feng, A.; Yue, J.; Li, L.; Wang, X.; Wang, X. The Efficacy and Safety of Regorafenib in Combination With Anti-PD-1 Antibody in Refractory Microsatellite Stable Metastatic Colorectal Cancer: A Retrospective Study. Front. Oncol. 2020, 10, 594125. [Google Scholar] [CrossRef]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Bendell, J.C.; Powderly, J.D.; Lieu, C.H.; Eckhardt, S.G.; Hurwitz, H.; Hochster, H.S.; Murphy, J.E.; Funke, R.P.; Rossi, C.; Wallin, J.; et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or FOLFOX in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 704. [Google Scholar] [CrossRef]
- Bendell, J.C.; Kim, T.W.; Goh, B.C.; Wallin, J.; Oh, D.-Y.; Han, S.-W.; Lee, C.B.; Hellmann, M.D.; Desai, J.; Lewin, J.H.; et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34, 3502. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Shiu, K.-K.; Andre, T.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177: Phase III randomized study of pembrolizumab versus chemotherapy for microsatellite instability-high advanced colorectal cancer. J. Clin. Oncol. 2021, 39, 6. [Google Scholar] [CrossRef]
- Andre, T.; Amonkar, M.; Norquist, J.M.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.; Garcia-Carbonero, R.; et al. Health-related quality of life in patients with microsatellite instability-high or mismatch repair deficient metastatic colorectal cancer treated with first-line pembrolizumab versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 665–677. [Google Scholar] [CrossRef]
- Shahda, S.; Noonan, A.M.; Bekaii-Saab, T.S.; O’Neil, B.H.; Sehdev, A.; Shaib, W.L.; Helft, P.R.; Loehrer, P.J.; Tong, Y.; Liu, Z.; et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. Presented at the American Society of Clinical Oncology 2017, Chicago, IL, USA, 2–6 June 2017. [Google Scholar]
- Ghiringhelli, F.; Fumet, J.D. Is There a Place for Immunotherapy for Metastatic Microsatellite Stable Colorectal Cancer? Front. Immunol. 2019, 10, 1816. [Google Scholar] [CrossRef] [Green Version]
- Schmoll, H.J.; Arnold, D.; de Gramont, A.; Ducreux, M.; Grothey, A.; O’Dwyer, P.J.; Van Cutsem, E.; Hermann, F.; Bosanac, I.; Bendahmane, B.; et al. MODUL-a multicenter randomized clinical trial of biomarker-driven maintenance therapy following first-line standard induction treatment of metastatic colorectal cancer: An adaptable signal-seeking approach. J. Cancer Res. Clin. Oncol. 2018, 144, 1197–1204. [Google Scholar] [CrossRef]
- Bendell, J.C.; Ciardiello, F.; Tabernero, J.; Tebbutt, N.; Eng, C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.; et al. Efficacy and safety results from IMblaze370, a randomised phase III study comparing atezolizumab plus cobimetinib and atezolizumab monotherapy vs. regorafenib in chemotherapy-refractory metastatic colorectal cancer. Presented at the ESMO World Congress on Gastrointestinal Cancer 2018, Barcelona, Spain, 20–23 June 2018. [Google Scholar]
- Hellmann, M.D.; Kim, T.W.; Lee, C.B.; Goh, B.C.; Miller, W.H., Jr.; Oh, D.Y.; Jamal, R.; Chee, C.E.; Chow, L.Q.M.; Gainor, J.F.; et al. Phase Ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann. Oncol. 2019, 30, 1134–1142. [Google Scholar] [CrossRef]
- Tabernero, J.; Melero, I.; Ros, W.; Argiles, G.; Marabelle, A.; Rodriguez-Ruiz, M.E.; Albanell, J.; Calvo, E.; Moreno, V.; Cleary, J.M.; et al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: Preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). Presented at the American Society of Clinical Oncology 2017, Chicago, IL, USA, 2–6 June 2017. [Google Scholar]
- Perez, R.P.; Riese, M.J.; Lewis, K.D.; Saleh, M.N.; Daud, A.; Berlin, J.; Lee, J.J.; Mukhopadhyay, S.; Zhou, L.; Serbest, G.; et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J. Clin. Oncol. 2017, 35, 3003. [Google Scholar] [CrossRef]
- Segal, N.H.; Kemeny, N.E.; Cercek, A.; Reidy, D.L.; Raasch, P.J.; Warren, P.; Hrabovsky, A.E.; Campbell, N.; Shia, J.; Goodman, K.A.; et al. Non- randomized phase II study to assess the efficacy of pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch repair proficient (pMMR) metastatic colorectal cancer (mCRC) patients. Presented at the American Society of Clinical Oncology 2016, Chicago, IL, USA, 3–7 June 2016. [Google Scholar]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. CD73: An emerging checkpoint for cancer immunotherapy. Immunotherapy 2019, 11, 983–997. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, D.A.; George, T.J., Jr.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chong, W.; Teng, C.; Yao, Y.; Wang, X.; Li, X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 2019, 110, 2348–2356. [Google Scholar] [CrossRef]
- Keenan, B.P.; Van Loon, K.; Khilnani, A.D.; Fidelman, N.; Behr, S.C.; Atreya, C.E.; Oh, D.Y. Molecular and Radiological Features of Microsatellite Stable Colorectal Cancer Cases With Dramatic Responses to Immunotherapy. Anticancer Res. 2021, 41, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jager, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 2019, 25, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Heij, L.R.; Grabsch, H.I.; Loeffler, C.; Echle, A.; Muti, H.S.; Krause, J.; Niehues, J.M.; Sommer, K.A.J.; Bankhead, P.; et al. Pan-cancer image-based detection of clinically actionable genetic alterations. Nat. Cancer 2020, 1, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackstadt, R.; van Hooff, S.R.; Leach, J.D.; Cortes-Lavaud, X.; Lohuis, J.O.; Ridgway, R.A.; Wouters, V.M.; Roper, J.; Kendall, T.J.; Roxburgh, C.S.; et al. Epithelial NOTCH Signaling Rewires the Tumor Microenvironment of Colorectal Cancer to Drive Poor-Prognosis Subtypes and Metastasis. Cancer Cell 2019, 36, 319–336.e7. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, K.P.; Loizou, E.; Livshits, G.; Schatoff, E.M.; Baslan, T.; Manchado, E.; Simon, J.; Romesser, P.B.; Leach, B.; Han, T.; et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat. Biotechnol. 2017, 35, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Accelerator Award, CRUK. Available online: https://www.cancerresearchuk.org/funding-for-researchers/accelerator-award/scientific-remit (accessed on 5 July 2021).
- Qiu, Q.; Lin, Y.; Ma, Y.; Li, X.; Liang, J.; Chen, Z.; Liu, K.; Huang, Y.; Luo, H.; Huang, R.; et al. Exploring the Emerging Role of the Gut Microbiota and Tumor Microenvironment in Cancer Immunotherapy. Front. Immunol. 2020, 11, 612202. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Classification | Name | Trade Name |
|---|---|---|
| Anti PD-1 | Pembrolizumab | Keytruda |
| Nivolumab | Opdivo | |
| Atezolizumab | Tecentriq | |
| Anti PD-L1 | Durvalumab | Imfinzi |
| Avelumab | Bavencio | |
| Anti CTLA4 | Ipilimumab | Yervoy |
| Tremelimumab | N/A |
| First Author | Journal | Year | Type | Target | Patient Selection | Generic | Type | Key Findings |
|---|---|---|---|---|---|---|---|---|
| Chung [47] | J Clin Oncol. | 2010 | Immune checkpoint inhibitor | CTLA4 | All patients | Tremelimumab | Phase II | Response rate 27% for nivolumab only, and 15% in nivolumab plus ipilimumab. |
| Topalian [48] | NEJM | 2012 | Immune checkpoint inhibitor | PD-1 | Includes NSCLC, MM, RCC, prostate ca and CRC. | BMS-936558 | Phase I | No clear benefit but one patient with partial response. |
| Brahmer [49] | NEJM | 2012 | Immune checkpoint inhibitor | PD-L1 | Includes CRC, RCC, ovarian ca, pancreatic ca, gastric ca, breast ca | Phase I | No objective responses in patients with CRC. | |
| Le [44] | NEJM | 2015 | Immune checkpoint inhibitor | PD-1 | Both dMMR and pMMR | Pembrolizumab | Phase II | Response rates at 31% by 12 months, with 69% disease control rate of 3 months or longer. |
| Bendell [55] | J Clin Oncol. | 2015 | Immune checkpoint inhibitor/bevacizumab or FOLFOX | PD-L1 | All patients | Atezolizumab | Phase 1b | No objective responses in patients with CRC. |
| Overman [46] | J Clin Oncol. | 2016 | Immune checkpoint inhibitor | PD-1/CTLA4 | All patients | Nivolumab and ipilimumab | Phase II | Adverse events occurred early, were manageable, and did not affect outcome |
| Bendell [56] | J Clin Oncol. | 2016 | Immune checkpoint inhibitor/MEK inhibitor | PD-L1/MEK | All patients | Atezolizumab | Phase 1b | Response rates were 8% for anti-PD-L1/bev, compared with 36% in patients with anti-PD-L1/bev/FOLFOX6 |
| Le [41] | Science | 2017 | Immune checkpoint inhibitor | PD-1 | High MSI/dMMR | Pembrolizumab | Phase II | Prolonged OS in advanced refractory CRC. |
| Overman [43] | Lancet Oncol. | 2017 | Immune checkpoint inhibitor | PD-1 | All patients | Nivolumab | Phase II | Response rates at 55%, with disease control rates for more than 3 months in 80%. |
| Overman [42] | J Clin Oncol. | 2018 | Immune checkpoint inhibitor | PD-1/ CTLA4 | High MSI/dMMR | Nivolumab and ipilimumab | Phase II | Responses in 53% patients, with complete responses in 21%. |
| Morse [45] | Oncologist | 2019 | Immune checkpoint inhibitor | PD-1/ CTLA4 | High MSI/dMMR | Nivolumab and ipilimumab | Phase II | Response rate 40% in dMMR and 0% in pMMR |
| Mettu [51] | Annals of Oncol. | 2019 | Immune checkpoint inhibitor/ capectabine/VEGF | PD-L1/ CTLA4 | All patients | Atezolizumab | Phase II | No patients had a tumour response. |
| Eng C [54] | Lancet Oncol. | 2019 | Immune checkpoint inhibitor/ MEK/VEGFR2 | PD-L1/ MEK/VEGFR2 | All patients | Atezolizumab | Phase III | No response, disease control rate in 78%, progression in 22%. |
| Antoniotti [39] | BMC Cancer | 2020 | Immune checkpoint inhibitor/ FOLFOXIRI/ bevacizumab | PD-L1 | All patients | Atezolizumab | Phase II | Combination strategy appears safe. Ongoing enrolment. |
| Chen [40] | JAMA Oncol. | 2020 | Immune checkpoint inhibitor | PD-L1/ CTLA4 | All patients | Durvalumab and Tremelimumab | Phase II | No major safety concerns. Ongoing enrolment. |
| Patel [50] | Cancer Medicine | 2020 | Immune checkpoint inhibitor/ trifluridine/tipiracil | PD-1 | MSS | Nivolumab | Phase II | Overall response rate 17%, not associated PD-L1 expression. |
| Li [53] | Frontiers in Oncol. | 2020 | Immune checkpoint inhibitor/RTK | PD-1/ VEGFR2 | MSS/pMMR | Mixture | Retrospective | Response rate 8%. |
| Andre [21] | NEJM | 2020 | Immune checkpoint inhibitor | PD-1 | MSI-H/dMMR | Pembrolizumab | Phase III | Response rate 44% vs. 33% (chemo), improved PFS. |
| Segal [52] | Clinical Cancer Res. | 2021 | Immune checkpoint inhibitor/RT | PD-L1/ CTLA4 | pMMR | Durvalumab and Tremelimumab | Phase II | Anti-PD-L1 added to capecitabine and bevacizumab improves response rates from 4 to 8%. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galbraith, N.J.; Wood, C.; Steele, C.W. Targeting Metastatic Colorectal Cancer with Immune Oncological Therapies. Cancers 2021, 13, 3566. https://doi.org/10.3390/cancers13143566
Galbraith NJ, Wood C, Steele CW. Targeting Metastatic Colorectal Cancer with Immune Oncological Therapies. Cancers. 2021; 13(14):3566. https://doi.org/10.3390/cancers13143566
Chicago/Turabian StyleGalbraith, Norman J., Colin Wood, and Colin W. Steele. 2021. "Targeting Metastatic Colorectal Cancer with Immune Oncological Therapies" Cancers 13, no. 14: 3566. https://doi.org/10.3390/cancers13143566
APA StyleGalbraith, N. J., Wood, C., & Steele, C. W. (2021). Targeting Metastatic Colorectal Cancer with Immune Oncological Therapies. Cancers, 13(14), 3566. https://doi.org/10.3390/cancers13143566