
Radiation Therapy in the Treatment of Head and Neck Rhabdomyosarcoma


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results and Discussion
2.1. Background
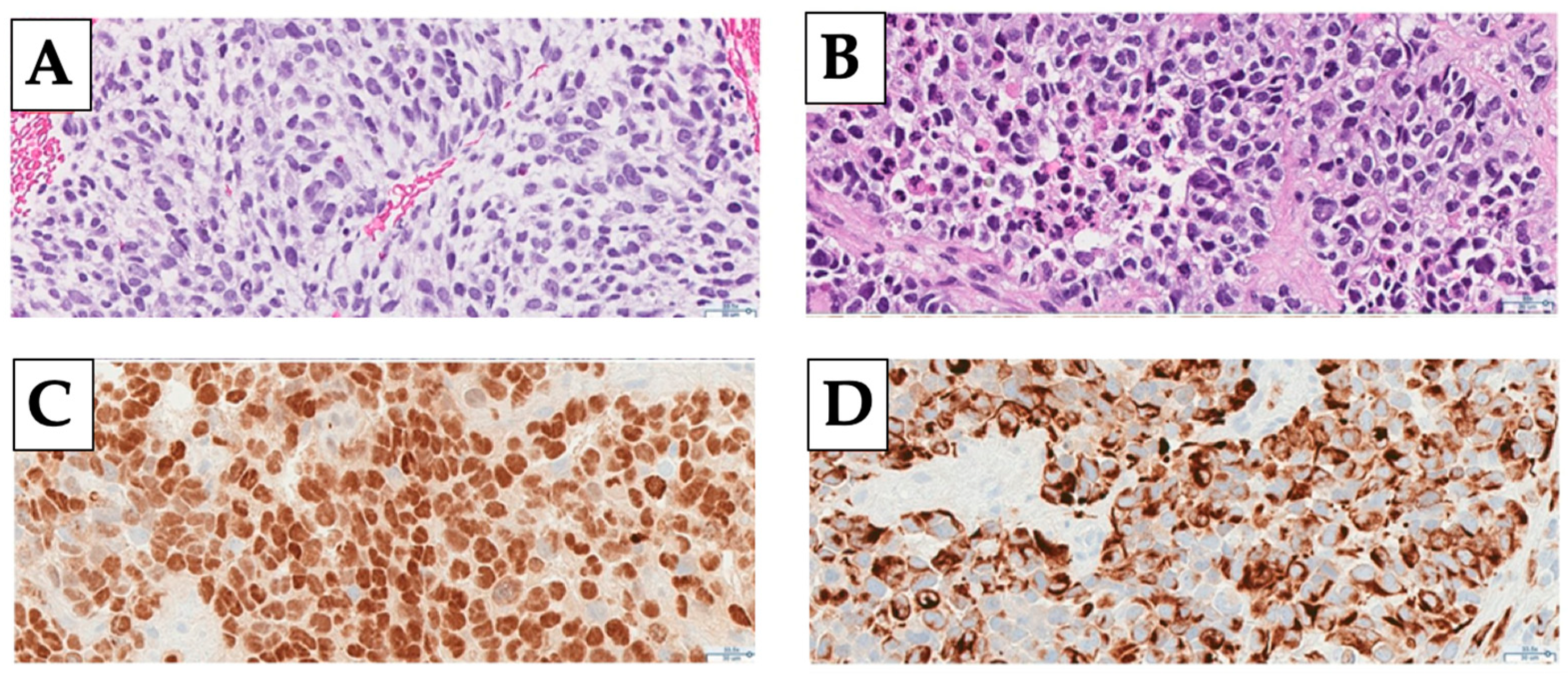
2.2. Diagnosis
2.3. Staging
2.4. Historical Rationale for Treatment Approach
2.4.1. Intergroup Rhabdomyosarcoma Studies
2.4.2. Children’s Oncology Group
2.5. Current Approach to Treatment in the Head and Neck
2.5.1. Surgery
2.5.2. Chemotherapy
2.5.3. Radiation Therapy
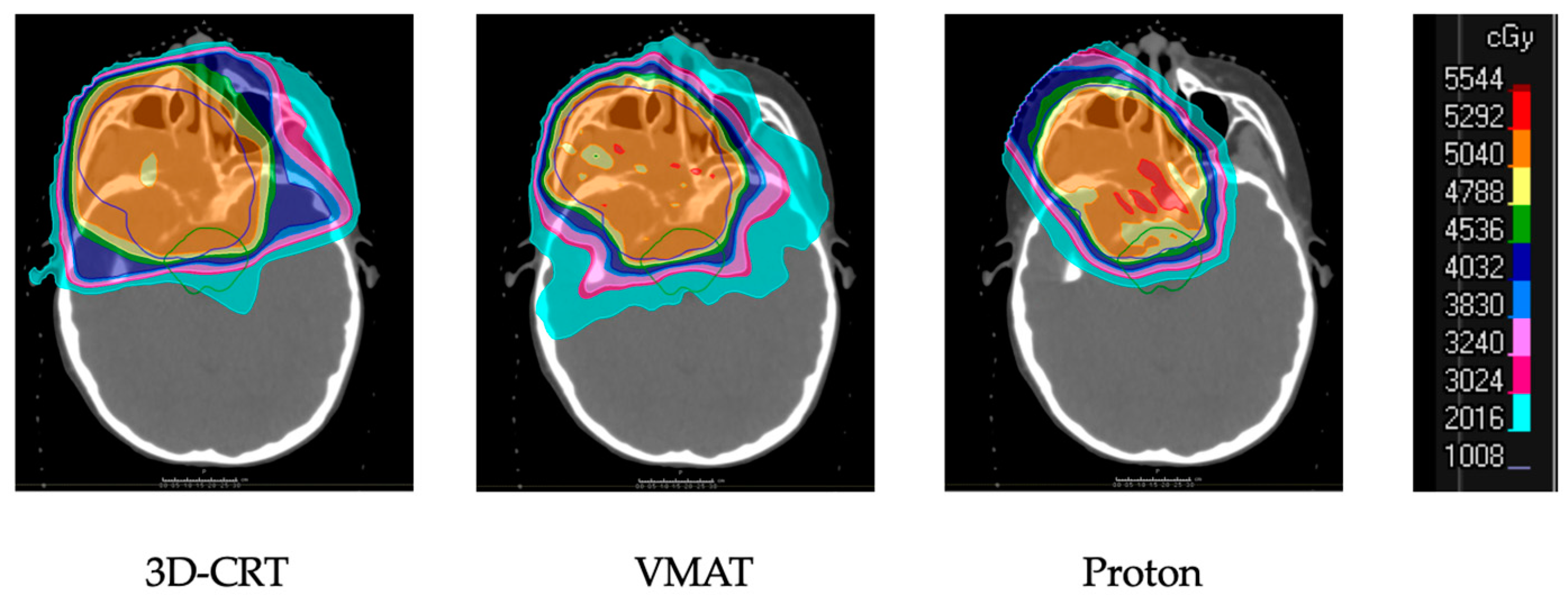
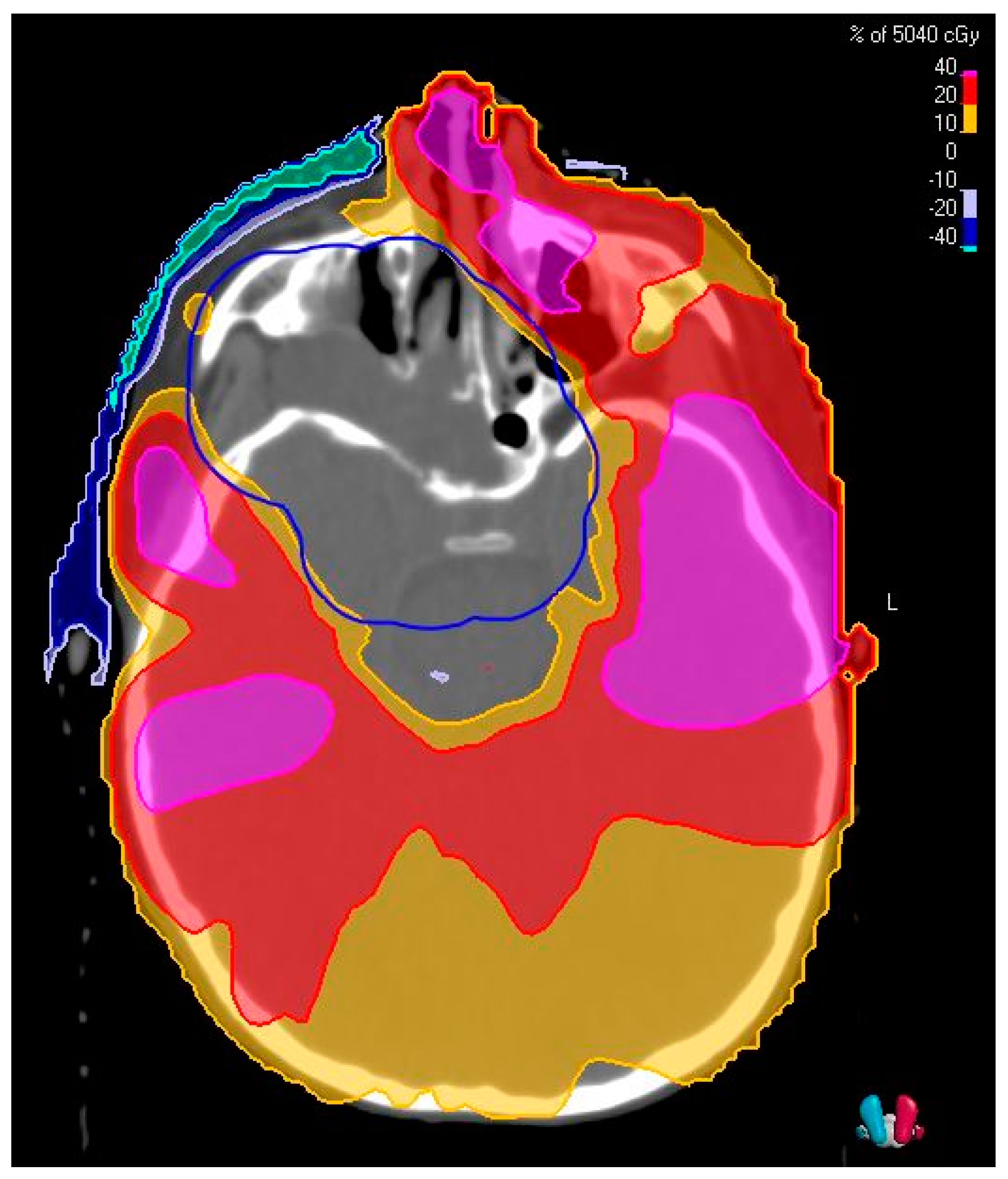
2.6. Overview of Techniques for Radiation Therapy
2.7. Considerations after Radiation Therapy
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Radzikowska, J.; Kukwa, W.; Kukwa, A.; Czarnecka, A.; Krzeski, A. Rhabdomyosarcoma of the Head and Neck in Children. Contemp. Oncol. 2015, 2, 98–107. [Google Scholar] [CrossRef]
- Terezakis, S.; Ladra, M. Pediatric Rhabdomyosarcoma. In Pediatric Radiation Oncology; Merchant, T.E., Kortmann, R.-D., Eds.; Pediatric Oncology; Springer International Publishing: Cham, Switzerland, 2018; pp. 21–43. [Google Scholar]
- Häußler, S.M.; Stromberger, C.; Olze, H.; Seifert, G.; Knopke, S.; Böttcher, A. Head and Neck Rhabdomyosarcoma in Children: A 20-Year Retrospective Study at a Tertiary Referral Center. J. Cancer Res. Clin. Oncol. 2018, 144, 371–379. [Google Scholar] [CrossRef]
- Blank, L.E.C.M.; Koedooder, K.; Pieters, B.R.; van der Grient, H.N.B.; van de Kar, M.; Buwalda, J.; Balm, A.J.M.; Merks, J.H.M.; Strackee, S.D.; Freling, N.J.; et al. The AMORE Protocol for Advanced-Stage and Recurrent Nonorbital Rhabdomyosarcoma in the Head-and-Neck Region of Children: A Radiation Oncology View. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1555–1562. [Google Scholar] [CrossRef]
- Carnevale, A.; Lieberman, E.; Cárdenas, R. Li-Fraumeni Syndrome in Pediatric Patients with Soft Tissue Sarcoma or Osteosarcoma. Arch. Med. Res. 1997, 28, 383–386. [Google Scholar] [PubMed]
- O’Neal, J.P.; Ramdas, J.; Wood, W.E.; Pellitteri, P.K. Parameningeal Rhabdomyosarcoma in a Patient with Costello Syndrome. J. Pediatr. Hematol. Oncol. 2004, 26, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Squire, J.A.; Thorner, P.; Zielenska, M.; Shuman, C.; Grant, R.; Chitayat, D.; Nishikawa, J.L.; Weksberg, R. Association of Alveolar Rhabdomyosarcoma with the Beckwith-Wiedemann Syndrome. Pediatr. Dev. Pathol. 2001, 4, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Pondrom, M.; Bougeard, G.; Karanian, M.; Bonneau-Lagacherie, J.; Boulanger, C.; Boutroux, H.; Briandet, C.; Chevreau, C.; Corradini, N.; Coze, C.; et al. Rhabdomyosarcoma Associated with Germline TP53 Alteration in Children and Adolescents: The French Experience. Pediatr. Blood Cancer 2020, 67. [Google Scholar] [CrossRef] [PubMed]
- Jawad, N.; McHugh, K. The Clinical and Radiologic Features of Paediatric Rhabdomyosarcoma. Pediatr. Radiol. 2019, 49, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup Rhabdomyosarcoma Study-IV: Results for Patients With Nonmetastatic Disease. JCO 2001, 19, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- Owosho, A.A.; Huang, S.-C.; Chen, S.; Kashikar, S.; Estilo, C.L.; Wolden, S.L.; Wexler, L.H.; Huryn, J.M.; Antonescu, C.R. A Clinicopathologic Study of Head and Neck Rhabdomyosarcomas Showing FOXO1 Fusion-Positive Alveolar and MYOD1 -Mutant Sclerosing Are Associated with Unfavorable Outcome. Oral. Oncol. 2016, 61, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Parham, D.M.; Qualman, S.J.; Teot, L.; Barr, F.G.; Morotti, R.; Sorensen, P.H.B.; Triche, T.J.; Meyer, W.H. Correlation Between Histology and PAX/FKHR Fusion Status in Alveolar Rhabdomyosarcoma: A Report From the Children’s Oncology Group. Am. J. Surg. Pathol. 2007, 31, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 Fusion Status Drives Unfavorable Outcome for Children with Rhabdomyosarcoma: A Children’s Oncology Group Report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR Gene Fusions Are Prognostic Indicators in Alveolar Rhabdomyosarcoma: A Report from the Children’s Oncology Group. JCO 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.; Lyden, E.R.; Anderson, J.R.; Hawkins, D.S.; Spunt, S.L.; Walterhouse, D.O.; Wolden, S.L.; Parham, D.M.; Rodeberg, D.A.; Kao, S.C.; et al. Histologic and Clinical Characteristics Can Guide Staging Evaluations for Children and Adolescents with Rhabdomyosarcoma: A Report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. J. Clin. Oncol. 2013, 31, 3226–3232. [Google Scholar] [CrossRef] [PubMed]
- Karanian, M.; Pissaloux, D.; Gomez-Brouchet, A.; Chevenet, C.; Le Loarer, F.; Fernandez, C.; Minard, V.; Corradini, N.; Castex, M.-P.; Duc-Gallet, A.; et al. SRF-FOXO1 and SRF-NCOA1 Fusion Genes Delineate a Distinctive Subset of Well-Differentiated Rhabdomyosarcoma. Am. J. Surg. Pathol. 2020, 44, 607–616. [Google Scholar] [CrossRef]
- Zorzi, A.P.; Grant, R.; Gupta, A.A.; Hodgson, D.C.; Nathan, P.C. Cranial Nerve Palsies in Childhood Parameningeal Rhabdomyosarcoma: CNP and PM RMS. Pediatr. Blood Cancer 2012, 59, 1211–1214. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, M.S.; Lee, B.H.; Choe, D.H.; Do, Y.S.; Kim, K.H.; Chin, S.Y.; Shim, Y.S.; Cho, K.J. Rhabdomyosarcoma of the Head and Neck in Adults: MR and CT Findings. AJNR Am. J. Neuroradiol. 1996, 17, 1923–1928. [Google Scholar]
- Mandell, L.R.; Massey, V.; Ghavimi, F. The Influence of Extensive Bone Erosion on Local Control in Non-Orbital Rhabdomyosarcoma of the Head and Neck. Int. J. Radiat. Oncol. Biol. Phys. 1989, 17, 649–653. [Google Scholar] [CrossRef]
- Freling, N.J.M.; Merks, J.H.M.; Saeed, P.; Balm, A.J.M.; Bras, J.; Pieters, B.R.; Adam, J.A.; van Rijn, R.R. Imaging Findings in Craniofacial Childhood Rhabdomyosarcoma. Pediatr. Radiol. 2010, 40, 1723–1738. [Google Scholar] [CrossRef] [Green Version]
- Pourmehdi Lahiji, A.; Jackson, T.; Nejadnik, H.; von Eyben, R.; Rubin, D.; Spunt, S.L.; Quon, A.; Daldrup-Link, H. Association of Tumor [18F]FDG Activity and Diffusion Restriction with Clinical Outcomes of Rhabdomyosarcomas. Mol. Imaging Biol. 2019, 21, 591–598. [Google Scholar] [CrossRef]
- Spunt, S.L.; Anderson, J.R.; Teot, L.A.; Breneman, J.C.; Meyer, W.H.; Pappo, A.S. Intergroup Rhabdomyosarcoma Study Group Routine Brain Imaging Is Unwarranted in Asymptomatic Patients with Rhabdomyosarcoma Arising Outside of the Head and Neck Region That Is Metastatic at Diagnosis: A Report from the Intergroup Rhabdomyosarcoma Study Group. Cancer 2001, 92, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Gradoni, P.; Giordano, D.; Oretti, G.; Fantoni, M.; Ferri, T. The Role of Surgery in Children with Head and Neck Rhabdomyosarcoma and Ewing’s Sarcoma. Surg. Oncol. 2010, 19, e103–e109. [Google Scholar] [CrossRef]
- Norman, G.; Fayter, D.; Lewis-Light, K.; Chisholm, J.; McHugh, K.; Levine, D.; Jenney, M.; Mandeville, H.; Gatz, S.; Phillips, B. An Emerging Evidence Base for PET-CT in the Management of Childhood Rhabdomyosarcoma: Systematic Review. BMJ Open 2015, 5, e006030. [Google Scholar] [CrossRef] [PubMed]
- Klem, M.L.; Grewal, R.K.; Wexler, L.H.; Schöder, H.; Meyers, P.A.; Wolden, S.L. PET for Staging in Rhabdomyosarcoma: An Evaluation of PET as an Adjunct to Current Staging Tools. J. Pediatric Hematol. Oncol. 2007, 29, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Chi, Y.; Tian, J.; Hingorani, P.; Mascarenhas, L.; McCowage, G.B.; Weigel, B.J.; Venkatramani, R.; Wolden, S.L.; Yock, T.I.; et al. Metabolic Response as Assessed by 18F-fluorodeoxyglucose Positron Emission Tomography-computed Tomography Does Not Predict Outcome in Patients with Intermediate- or High-risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. Cancer Med. 2021, 10, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Rodeberg, D.A.; Garcia-Henriquez, N.; Lyden, E.R.; Davicioni, E.; Parham, D.M.; Skapek, S.X.; Hayes-Jordan, A.A.; Donaldson, S.S.; Brown, K.L.; Triche, T.J.; et al. Prognostic Significance and Tumor Biology of Regional Lymph Node Disease in Patients With Rhabdomyosarcoma: A Report From the Children’s Oncology Group. JCO 2011, 29, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.; Pressey, J.G.; Nagarajan, R.; Weiss, B.D.; Trout, A.T.; Gelfand, M.J.; Pater, L.; Vatner, R.E.; Breneman, J.C.; Dasgupta, R. Sentinel Lymph Node Biopsy in Head and Neck Rhabdomyosarcoma. Pediatr Blood Cancer 2019, 66, e27532. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.S.; Castro, J.R.; Wilbur, J.R.; Jesse, R.H. Rhabdomyosarcoma of Head and Neck in Children. Combination Treatment by Surgery, Irradiation, and Chemotherapy. Cancer 1973, 31, 26–35. [Google Scholar] [CrossRef]
- Stobbe, G.D.; Dargeon, H.W. Embryonal Rhabdomyosarcoma of the Head and Neck in Children and Adolescents. Cancer 1950, 3, 826–836. [Google Scholar] [CrossRef]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; Wharam, M.D.; Wiener, E.S.; Crist, W.M. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major Lessons from the IRS-I through IRS-IV Studies as Background for the Current IRS-V Treatment Protocols. Sarcoma 2001, 5, 9–15. [Google Scholar] [CrossRef]
- Maurer, H.M.; Beltangady, M.; Gehan, E.A.; Crist, W.; Hammond, D.; Hays, D.M.; Heyn, R.; Lawrence, W.; Newton, W.; Ortega, J. The Intergroup Rhabdomyosarcoma Study-I. A Final Report. Cancer 1988, 61, 209–220. [Google Scholar] [CrossRef]
- Maurer, H.M.; Gehan, E.A.; Beltangady, M.; Crist, W.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J. The Intergroup Rhabdomyosarcoma Study-II. Cancer 1993, 71, 1904–1922. [Google Scholar] [CrossRef]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; Heyn, R. The Third Intergroup Rhabdomyosarcoma Study. J. Clin. Oncol. 1995, 13, 610–630. [Google Scholar] [CrossRef]
- Donaldson, S.S.; Meza, J.; Breneman, J.C.; Crist, W.M.; Laurie, F.; Qualman, S.J.; Wharam, M. Children’s Oncology Group Soft Tissue Sarcoma Committee (formely Intergroup Rhabdomyosarcoma Group) representing the Children’s Oncology Group and the Quality Assurance Review Center Results from the IRS-IV Randomized Trial of Hyperfractionated Radiotherapy in Children with Rhabdomyosarcoma--a Report from the IRSG. Int J. Radiat. Oncol. Biol. Phys. 2001, 51, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Breneman, J.C.; Donaldson, S.S.; Constine, L.; Merchant, T.; Marcus, K.; Paulino, A.C.; Followill, D.; Mahajan, A.; Laack, N.; Esiashvili, N.; et al. The Children’s Oncology Group Radiation Oncology Discipline: 15 Years of Contributions to the Treatment of Childhood Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 101, 860–874. [Google Scholar] [CrossRef]
- Beverly Raney, R.; Walterhouse, D.O.; Meza, J.L.; Andrassy, R.J.; Breneman, J.C.; Crist, W.M.; Maurer, H.M.; Meyer, W.H.; Parham, D.M.; Anderson, J.R. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 Protocol, Using Vincristine and Dactinomycin with or without Cyclophosphamide and Radiation Therapy, for Newly Diagnosed Patients with Low-Risk Embryonal Rhabdomyosarcoma: A Report From the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. JCO 2011, 29, 1312–1318. [Google Scholar] [CrossRef]
- Breneman, J.; Meza, J.; Donaldson, S.S.; Raney, R.B.; Wolden, S.; Michalski, J.; Laurie, F.; Rodeberg, D.A.; Meyer, W.; Walterhouse, D.; et al. Local Control with Reduced-Dose Radiotherapy for Low-Risk Rhabdomyosarcoma: A Report from the Children’s Oncology Group D9602 Study. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Shorter-Duration Therapy Using Vincristine, Dactinomycin, and Lower-Dose Cyclophosphamide with or without Radiotherapy for Patients with Newly Diagnosed Low-Risk Rhabdomyosarcoma: A Report From the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. JCO 2014, 32, 3547–3552. [Google Scholar] [CrossRef] [Green Version]
- Ermoian, R.P.; Breneman, J.; Walterhouse, D.O.; Chi, Y.-Y.; Meza, J.; Anderson, J.; Hawkins, D.S.; Hayes-Jordan, A.A.; Parham, D.M.; Yock, T.I.; et al. 45 Gy is Not Sufficient Radiotherapy Dose for Group III Orbital Embryonal Rhabdomyosarcoma after Less than Complete Response to 12 Weeks of ARST0331 Chemotherapy: A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64, e26540. [Google Scholar] [CrossRef]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Reduction of Cyclophosphamide Dose for Patients with Subset 2 Low-Risk Rhabdomyosarcoma Is Associated with an Increased Risk of Recurrence: A Report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group: Lower Cyclophosphamide for Rhabdomyosarcoma. Cancer 2017, 123, 2368–2375. [Google Scholar] [CrossRef]
- Arndt, C.A.S.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, Actinomycin, and Cyclophosphamide Compared with Vincristine, Actinomycin, and Cyclophosphamide Alternating with Vincristine, Topotecan, and Cyclophosphamide for Intermediate-Risk Rhabdomyosarcoma: Children’s Oncology Group Study D9803. JCO 2009, 27, 5182–5188. [Google Scholar] [CrossRef] [Green Version]
- Wolden, S.L.; Lyden, E.R.; Arndt, C.A.; Hawkins, D.S.; Anderson, J.R.; Rodeberg, D.A.; Morris, C.D.; Donaldson, S.S. Local Control for Intermediate-Risk Rhabdomyosarcoma: Results From D9803 According to Histology, Group, Site, and Size: A Report From the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; Donaldson, S.S.; Meza, J.L.; Anderson, J.R.; Lyden, E.R.; Brown, C.K.; Morano, K.; Laurie, F.; Arndt, C.A.; Enke, C.A.; et al. Effect of Radiotherapy Techniques (IMRT vs. 3D-CRT) on Outcome in Patients with Intermediate-Risk Rhabdomyosarcoma Enrolled in COG D9803—A Report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1764–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, D.L.; Chi, Y.; Donaldson, S.S.; Hawkins, D.S.; Tian, J.; Arndt, C.A.; Rodeberg, D.A.; Routh, J.C.; Lautz, T.B.; Gupta, A.A.; et al. Increased Local Failure for Patients with Intermediate-risk Rhabdomyosarcoma on ARST0531: A Report from the Children’s Oncology Group. Cancer 2019, 125, 3242–3248. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination with Chemotherapy for First Relapse Rhabdomyosarcoma: A Report from the Children’s Oncology Group. JCO 2019, 37, 2866–2874. [Google Scholar] [CrossRef]
- Lautz, T.B.; Chi, Y.; Li, M.; Wolden, S.L.; Casey, D.L.; Routh, J.C.; Granberg, C.F.; Binite, O.; Rudzinski, E.R.; Hawkins, D.S.; et al. Benefit of Delayed Primary Excision in Rhabdomyosarcoma: A Report from the Children’s Oncology Group. Cancer 2021, 127, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.L.; Wexler, L.H.; Wolden, S.L. Worse Outcomes for Head and Neck Rhabdomyosarcoma Secondary to Reduced-Dose Cyclophosphamide. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 1151–1157. [Google Scholar] [CrossRef]
- Spalding, A.C.; Hawkins, D.S.; Donaldson, S.S.; Anderson, J.R.; Lyden, E.; Laurie, F.; Wolden, S.L.; Arndt, C.A.S.; Michalski, J.M. The Effect of Radiation Timing on Patients with High-Risk Features of Parameningeal Rhabdomyosarcoma: An Analysis of IRS-IV and D9803. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Ladra, M.; Marcus, K.J.; Yock, T. Rhabdomyosarcoma. In Target Volume Delineation for Pediatric Cancers; Terezakis, S.A., MacDonald, S.M., Eds.; Practical Guides in Radiation Oncology; Springer International Publishing: Cham, Switzerland, 2019; pp. 125–144. [Google Scholar]
- Laskar, S.; Pandit, P.; Mallik, S.; Tike, P.; Chaudhari, S.; Khanna, N.; Vora, T. Adaptive Radiation Therapy for Pediatric Head and Neck Malignancies: Dosimetric Implications. Pract. Radiat. Oncol. 2015, 5, e87–e94. [Google Scholar] [CrossRef]
- Veldeman, L.; Madani, I.; Hulstaert, F.; De Meerleer, G.; Mareel, M.; De Neve, W. Evidence behind Use of Intensity-Modulated Radiotherapy: A Systematic Review of Comparative Clinical Studies. Lancet Oncol. 2008, 9, 367–375. [Google Scholar] [CrossRef]
- Miralbell, R.; Lomax, A.; Cella, L.; Schneider, U. Potential Reduction of the Incidence of Radiation-Induced Second Cancers by Using Proton Beams in the Treatment of Pediatric Tumors. Int J. Radiat. Oncol. Biol. Phys. 2002, 54, 824–829. [Google Scholar] [CrossRef]
- Nguyen, F.; Rubino, C.; Guerin, S.; Diallo, I.; Samand, A.; Hawkins, M.; Oberlin, O.; Lefkopoulos, D.; De Vathaire, F. Risk of a Second Malignant Neoplasm after Cancer in Childhood Treated with Radiotherapy: Correlation with the Integral Dose Restricted to the Irradiated Fields. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Both, S.; Kirk, M.; Chao, H.-H.; Bagatell, R.; Li, Y.; Womer, R.; Balamuth, N.; Reilly, A.; Kurtz, G.; et al. Proton Therapy for Pediatric Head and Neck Malignancies. Pediatr. Blood Cancer 2018, 65, e26858. [Google Scholar] [CrossRef]
- De, B.; Kinnaman, M.D.; Wexler, L.H.; Kramer, K.; Wolden, S.L. Central Nervous System Relapse of Rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26710. [Google Scholar] [CrossRef] [PubMed]
- de Mattos, V.D.; Ferman, S.; Magalhães, D.M.A.; Antunes, H.S.; Lourenço, S.Q.C. Dental and Craniofacial Alterations in Long-Term Survivors of Childhood Head and Neck Rhabdomyosarcoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 127, 272–281. [Google Scholar] [CrossRef]
- Owosho, A.A.; Brady, P.; Wolden, S.L.; Wexler, L.H.; Antonescu, C.R.; Huryn, J.M.; Estilo, C.L. Long-Term Effect of Chemotherapy–Intensity-Modulated Radiation Therapy (Chemo-IMRT) on Dentofacial Development in Head and Neck Rhabdomyosarcoma Patients. Pediatric Hematol. Oncol. 2016, 33, 383–392. [Google Scholar] [CrossRef]
- Schoot, R.A.; Hol, M.L.F.; Merks, J.H.M.; Suttie, M.; Slater, O.; van Lennep, M.; Hopman, S.M.J.; Dunaway, D.; Syme-Grant, J.; Smeele, L.E.; et al. Facial Asymmetry in Head and Neck Rhabdomyosarcoma Survivors: SCHOOT et al. Pediatr. Blood Cancer 2017, 64, e26508. [Google Scholar] [CrossRef] [PubMed]
- Lockney, N.A.; Friedman, D.N.; Wexler, L.H.; Sklar, C.A.; Casey, D.L.; Wolden, S.L. Late Toxicities of Intensity-Modulated Radiation Therapy for Head and Neck Rhabdomyosarcoma. Pediatr. Blood Cancer 2016, 63, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Vaarwerk, B.; Schoot, R.A.; Maurice-Stam, H.; Slater, O.; Hartley, B.; Saeed, P.; Gajdosova, E.; van den Brekel, M.W.; Balm, A.J.M.; Hol, M.L.F.; et al. Psychosocial Well-Being of Long-Term Survivors of Pediatric Head-Neck Rhabdomyosarcoma. Pediatr. Blood Cancer 2019, 66, e27498. [Google Scholar] [CrossRef]
- Boutroux, H.; Levy, C.; Mosseri, V.; Desjardins, L.; Plancher, C.; Helfre, S.; Freneaux, P.; Cellier, C.; Orbach, D. Long-Term Evaluation of Orbital Rhabdomyosarcoma in Children: Orbital Rhabdomyosarcoma in Children. Clin. Exp. Ophthalmol 2015, 43, 12–19. [Google Scholar] [CrossRef]
- Raney, R.B.; Anderson, J.R.; Kollath, J.; Vassilopoulou-Sellin, R.; Klein, M.J.; Heyn, R.; Glicksman, A.S.; Wharam, M.; Crist, W.M.; Maurer, H.M. Late Effects of Therapy in 94 Patients with Localized Rhabdomyosarcoma of the Orbit: Report from the Intergroup Rhabdomyosarcoma Study (IRS)-III, 1984-1991. Med. Pediatr. Oncol. 2000, 34, 413–420. [Google Scholar] [CrossRef]
- Ludmir, E.B.; Grosshans, D.R.; McAleer, M.F.; McGovern, S.L.; Harrison, D.J.; Okcu, M.F.; Chintagumpala, M.M.; Mahajan, A.; Paulino, A.C. Patterns of Failure Following Proton Beam Therapy for Head and Neck Rhabdomyosarcoma. Radiother. Oncol. 2019, 134, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Vatner, R.E.; Niemierko, A.; Misra, M.; Weyman, E.A.; Goebel, C.P.; Ebb, D.H.; Jones, R.M.; Huang, M.S.; Mahajan, A.; Grosshans, D.R.; et al. Endocrine Deficiency as a Function of Radiation Dose to the Hypothalamus and Pituitary in Pediatric and Young Adult Patients with Brain Tumors. J. Clin. Oncol. 2018, 36, 2854–2862. [Google Scholar] [CrossRef] [PubMed]
- Darzy, K.H. Radiation-Induced Hypopituitarism after Cancer Therapy: Who, How and When to Test. Nat. Clin. Pract. Endocrinol. Metab. 2009, 5, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Renaux, V.K.; Bulteau, C.; Viguier, D.; Levy-Piebois, C.; Sainte-Rose, C.; Dellatolas, G.; Raquin, M.-A.; Jambaqué, I.; Kalifa, C. Long-Term Intellectual Outcome in Children with Posterior Fossa Tumors According to Radiation Doses and Volumes. Int. J. Radiat. Oncol. Biol. Phys. 1999, 45, 137–145. [Google Scholar] [CrossRef]
- Mahajan, A.; Stavinoha, P.L.; Rongthong, W.; Brodin, N.P.; McGovern, S.L.; El Naqa, I.; Palmer, J.D.; Vennarini, S.; Indelicato, D.J.; Aridgides, P.; et al. Neurocognitive Effects and Necrosis in Childhood Cancer Survivors Treated with Radiation Therapy: A PENTEC Comprehensive Review. Int. J. Radiat. Oncol. Biol. Phys. 2021, S0360301621001279. [Google Scholar] [CrossRef]
- Cohen, R.J.; Curtis, R.E.; Inskip, P.D.; Fraumeni, J.F. The Risk of Developing Second Cancers among Survivors of Childhood Soft Tissue Sarcoma. Cancer 2005, 103, 2391–2396. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Site | Tumor Size | Nodal Involvement | Metastatic Spread |
|---|---|---|---|---|
| I | Favorable | Any | Any | No |
| II | Unfavorable | ≤5 cm | No | No |
| III | Unfavorable | ≤5 cm >5 cm | Yes Any | No No |
| IV | Any | Any | Any | Yes |
| IRSG Grouping | Description |
|---|---|
| Group I | Localized disease with microscopic margin-negative (R0) resection A–Confined to muscle/organ of presentation B–Extension beyond muscle/organ of presentation |
| Group II | Gross total resection A–Margin-positive resection (R1) B–Regional lymph node involvement with R0 resection of nodal disease C–Regional lymph node involvement with R1 resection of nodal disease |
| Group III | Gross residual disease following resection A–Biopsy only B–>50% resection completed |
| Group IV | Distant metastatic disease at presentation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frankart, A.J.; Breneman, J.C.; Pater, L.E. Radiation Therapy in the Treatment of Head and Neck Rhabdomyosarcoma. Cancers 2021, 13, 3567. https://doi.org/10.3390/cancers13143567
Frankart AJ, Breneman JC, Pater LE. Radiation Therapy in the Treatment of Head and Neck Rhabdomyosarcoma. Cancers. 2021; 13(14):3567. https://doi.org/10.3390/cancers13143567
Chicago/Turabian StyleFrankart, Andrew J., John C. Breneman, and Luke E. Pater. 2021. "Radiation Therapy in the Treatment of Head and Neck Rhabdomyosarcoma" Cancers 13, no. 14: 3567. https://doi.org/10.3390/cancers13143567
APA StyleFrankart, A. J., Breneman, J. C., & Pater, L. E. (2021). Radiation Therapy in the Treatment of Head and Neck Rhabdomyosarcoma. Cancers, 13(14), 3567. https://doi.org/10.3390/cancers13143567